Denne vejledning indeholder information til fabrikanter af medicinsk software og definerer kriterierne for kvalifikation af software. Vejledningen indeholder information om anvendelsen af klassificeringskriterier for software i henhold til forordning om medicinsk udstyr (EU) 2017/745 (MDR) og forordning om medicinsk udstyr til in vitro-diagnostik (EU) 2017/746 (IVDR) samt information relateret til markedsføring.
Kriterierne specificeret i denne vejledning gælder også for applikationer (ofte benævnt apps), som kan fungere på en mobiltelefon, i skyen eller på andre platforme.
Lovgivningen om medicinsk udstyr er opdelt i 2 hovedkategorier af medicinsk udstyr – 1) medicinsk udstyr og 2) medicinsk udstyr til in vitro-diagnostik.
Medicinsk udstyr omfatter også produkter uden et medicinsk formål, der er nævnt på listen i bilag XVI i forordningen om medicinsk udstyr (EU) 2017/745 (MDR).
De EU-retslige regler om medicinsk udstyr bliver den 26. maj 2021 erstattet af Europa-Parlamentets og Rådets forordning (EU) 2017/745 af 5. april 2017 om medicinsk udstyr. Forordningen finder direkte anvendelse i Danmark.
De EU-retslige regler om medicinsk udstyr til in vitro-diagnostik bliver erstattet af Europa-Parlamentets og Rådets forordning (EU) 2017/746 af 5. april 2017 om medicinsk udstyr til in vitro-diagnostik. Forordningen finder direkte anvendelse i Danmark.
Afsnit i denne vejledning, som omhandler medicinsk udstyr til in vitro-diagnostik finder først anvendelse, når forordningen om medicinsk udstyr til in vitro-diagnostik finder anvendelse. Dette gælder særligt for afsnit: 2.5, 2.6, 4 samt eksempler på kvalifikation og klassifikation af software som medicinsk udstyr til in vitro-diagnostik.
Vejledningen præciserer reglerne i Europa-Parlamentets og Rådets forordning (EU) 2017/745 af 5. april 2017 om medicinsk udstyr, Europa-Parlamentets og Rådets forordning (EU) 2017/746 af 5. april 2017 om medicinsk udstyr til in vitro-diagnostik. Vejledningen erstatter vejledning nr. 9865 af 2. september 2016 til fabrikanter om apps og software som medicinsk udstyr, som hermed ophæves.
Forordningen om medicinsk udstyr omtales i denne vejledning som ”MDR” og forordningen om medicinsk udstyr til in vitro-diagnostik omtales i denne vejledning som ”IVDR”. Se henvisninger til lovgivning i afsnit 15 i denne vejledning.
Indhold
| 1. | Definitioner og forkortelser | |
|---|---|---|
| 2. | Kvalifikation som medicinsk udstyr-software | |
| 2.1 | Introduktion til kvalifikationskriterier | |
| 2.2 | Medicinsk udstyr-software (MDSW) | |
| 2.3 | Software der styrer eller påvirker brugen af medicinsk udstyr | |
| 2.4 | Beslutningstrin til kvalificering af software som et MDSW (Figur 1) | |
| 2.5 | Kvalifikationskriterier for MDSW til in vitro-diagnostik | |
| 2.6 | Beslutningstrin for kvalificering af MDSW som medicinsk udstyr eller in vitro-diagnostisk medicinsk udstyr (Figur 2) | |
| 3. | Klassificering af MDSW jf. MDR | |
| 3.1 | Gennemførelsesbestemmelser | |
| 3.2 | Klassificeringsregler | |
| 4. | Klassificering- og gennemførelsesregler jf. IVDR | |
| 4.1 | Gennemførelsesregler | |
Medicinsk udstyr:
Ifølge Forordning (EU) 2017/745 – MDR og Forordning (EU) 2017/746 – IVDR, betyder ”Medicinsk udstyr” ethvert instrument, apparat, udstyr, software, implantat, reagens, materiale eller anden genstand, som ifølge fabrikanten er bestemt til anvendelse, alene eller i kombination, på mennesker med henblik på et eller flere af følgende særlige medicinske formål:
— diagnosticering, forebyggelse, monitorering, forudsigelse, prognose, behandling eller lindring af sygdomme
— diagnosticering, monitorering, behandling, afhjælpning af eller kompensation for skader eller handicap — afprøvning, udskiftning eller ændring af anatomien eller en fysiologisk eller patologisk proces eller tilstand
— tilvejebringelse af oplysninger ved hjælp af in vitro-undersøgelse af prøvemateriale fra det menneskelige legeme, herunder organ-, blod- og vævsdonationer, hvis forventede hovedvirkning i eller på det menneskelige legeme ikke fremkaldes ad farmakologisk, immunologisk eller metabolisk vej, men hvis virkning kan understøttes ad denne vej. Følgende produkter anses også for at være medicinsk udstyr:
— udstyr til svangerskabsforebyggelse eller -støtte
— produkter, der specifikt er beregnet til rengøring, desinfektion eller sterilisering af udstyr som omhandlet i MDR artikel 1, stk. 4, og af udstyr som omhandlet i dette nummers første afsnit.1
Medicinsk udstyr til in vitro-diagnostik:
Ifølge Forordning (EU) 2017/746 – IVDR, betyder ”medicinsk udstyr til in vitro-diagnostik” ethvert medicinsk udstyr, som er et reagens, et reagensprodukt, en kalibrator, et kontrolmateriale, et prøvesæt, et instrument, et apparat, en anordning, software eller et system, anvendt alene eller i kombination, og som ifølge fabrikanten er bestemt til anvendelse in vitro til undersøgelse af prøvemateriale fra det menneskelige legeme, herunder blod- og vævsdonationer, udelukkende eller hovedsagelig med henblik på at tilvejebringe oplysninger om en eller flere af følgende:
a) om en fysiologisk eller patologisk proces eller tilstand
b) om medfødte fysiske eller mentale handicap
c) om disposition for en medicinsk tilstand eller sygdom
d) til fastlæggelse af sikkerhed for og kompatibilitet med potentielle recipienter
e) til forudsigelse af reaktioner på behandlinger
f) til definition eller monitorering af terapeutiske foranstaltninger.
Prøvebeholdere anses også for at være medicinsk udstyr til in vitro-diagnostik.2
Tilbehør til medicinsk udstyr:
Ifølge Forordning (EU) 2017/745 – MDR, betyder ”Tilbehør til medicinsk udstyr” enhver genstand, der, selv om den ikke i sig selv er medicinsk udstyr, ifølge fabrikanten er bestemt til at blive anvendt sammen med en eller flere slags bestemt medicinsk udstyr, for specifikt at muliggøre, at det medicinske udstyr kan anvendes i overensstemmelse med sit erklærede formål, eller for specifikt og direkte at hjælpe det medicinske udstyrs medicinske funktion i henhold til dets erklærede formål3.
Ifølge Forordning (EU) 2017/746 – IVDR, betyder ”Tilbehør til medicinsk udstyr” ethvert produkt, der, selv om det ikke i sig selv er medicinsk udstyr til in vitro-diagnostik, ifølge fabrikanten er bestemt til at blive anvendt sammen med en eller flere slags bestemt medicinsk udstyr til in vitro-diagnostik for specifikt at muliggøre, at dette kan anvendes i overensstemmelse med sit erklærede formål, eller for specifikt og direkte at hjælpe den medicinske funktion af det medicinske udstyr til in vitro-diagnostik i henhold til dets erklærede formål.4
Note: Software, som er tilbehør til medicinsk udstyr, kan være styrende eller påvirke brugen af et medicinsk udstyr.
Note: Fabrikanter skal i den tekniske dokumentation beskrive hvilke tilbehør, der er tiltænkt at blive anvendt i kombination med udstyret. Disse informationer skal også fremgå af brugsvejledningen til medicinsk udstyr eller in vitro-diagnostisk medicinsk udstyr, der muliggør anvendelse af dertilhørende software og tilbehør. 5
Aktivt udstyr:
Ifølge Forordning (EU) 2017/745 – MDR, betyder ”aktivt udstyr” ethvert udstyr, som for at kunne fungere er afhængigt af en anden form for energi end den, der udvikles af det menneskelige legeme til dette formål, eller tyngdekraften, og som virker ved at ændre densiteten af eller ved at omsætte denne energi. Udstyr, der er beregnet til uden nogen væsentlig ændring at overføre energi, stoffer eller andre elementer mellem aktivt udstyr og patienten, anses ikke for at være aktivt udstyr. Software anses også for at være aktivt udstyr.6
Erklæret formål:
Ifølge Forordning (EU) 2017/745 – MDR, betyder ”Erklæret formål”, den anvendelse, som et udstyr er bestemt til ifølge fabrikantens oplysninger på mærkningen, ifølge brugsanvisningen eller ifølge salgsfremmende materiale- eller salgsmateriale eller reklame- og salgserklæringer og som specificeret af fabrikanten i den kliniske evaluering.7
Ifølge Forordning (EU) 2017/746 – IVDR, betyder ”Erklæret formål”, den anvendelse, som et udstyr er bestemt til ifølge fabrikantens oplysninger på mærkningen, ifølge brugsanvisningen eller ifølge salgsfremmende materiale eller salgsmateriale eller reklame- og salgserklæringerne eller som specificeret af fabrikanten i ydeevneevalueringen.8
Gøre tilgængelig på markedet:
Ifølge Forordning (EU) 2017/745 – MDR, betyder ”Gøre tilgængelig på markedet” enhver levering af udstyr, bortset fra udstyr bestemt til afprøvning, med henblik på distribution, forbrug eller anvendelse på EU-markedet som led i erhvervsvirksomhed mod eller uden vederlag.9
Ifølge Forordning (EU) 2017/746 – IVDR, betyder ”Gøre tilgængelig på markedet” enhver levering af udstyr, bortset fra udstyr, hvis ydeevne skal undersøges, med henblik på distribution, forbrug eller anvendelse på EU-markedet som led i erhvervsvirksomhed mod eller uden vederlag.10
Ibrugtagning:
Ifølge Forordning (EU) 2017/745 – MDR, betyder ”Ibrugtagning” det stadium, hvor et udstyr, bortset fra udstyr bestemt til afprøvning, stilles til rådighed for slutbrugeren og er klar til for første gang at blive anvendt i overensstemmelse med sit erklærede formål på EU-markedet.11
Ifølge Forordning (EU) 2017/746 – IVDR, betyder ”Ibrugtagning” det stadium, hvor udstyr, bortset fra udstyr, hvis ydeevne skal undersøges, stilles til rådighed for slutbrugeren og er klar til for første gang at blive anvendt i overensstemmelse med sit erklærede formål på EU-markedet.12
Bemyndiget organ:
Ifølge Forordning (EU) 2017/745 – MDR og Forordning (EU) 2017/746 – IVDR, betyder ”Bemyndiget organ” et overensstemmelsesvurderingsorgan, der er udpeget i overensstemmelse med denne forordning. Overensstemmelsesvurderingsorgan er defineret som et organ, der som tredjepart udfører overensstemmelsesvurderingsopgaver, herunder kalibrering, testning, certificering og inspektion.13
Software:
Med henblik på denne vejledning, er ”software” defineret som et sæt af instruktioner, der behandler input data og skaber output data.
Input data:
Data der tilføres software for at skabe output data efter behandling af denne data, kan anses som input data. Eksempler på input data er (ikke udtømmende):
• Data genereret gennem brug af menneske data-input udstyr såsom tastatur, mus, pen eller touchskærm.
• Data givet gennem stemmegenkendelse.
• Digitalt dokument: formateret til generelle formål såsom en Word-fil eller et JPEG-billede, formateret til medicinske formål såsom en DICOM-fil eller EKG-målinger eller en Elektronisk Patient Journal eller et uformateret dokument. Bemærk at digitale dokumenter skal differentieres fra software, der kan læse sådanne dokumenter.
• Data modtaget fra/transmitteret af udstyr.
Output data:
Data, der produceres af software, kan betragtes som output data. Eksempler på output data er (ikke udtømmende):
• Skærmvisningsdata (såsom layout med tal, tegn, billeder, grafik og lign.).
• Fysisk printet data (såsom layout med tal, tegn, billeder, grafik og lign.).
• Lyddata.
• Digitalt dokument (formateret til generelle formål såsom en Word-fil eller et JPEG-billede, formateret til medicinske formål såsom en DICOM-fil eller EKG-målinger eller en Elektronisk Patient Journal eller et uformateret dokument.).
• Haptisk rysten som et alternativ til lyd.
Software der styrer eller påvirker brugen af udstyr:
Software hvis erklærede formål er at styre eller påvirke brugen af (hardware) medicinsk udstyr, og som ikke har eller udfører et medicinsk formål selvstændigt , eller skaber information uafhængigt til et eller flere medicinske formål, som beskrevet i definitionen af medicinsk udstyr eller medicinsk udstyr til in vitro-diagnostik. Denne software kan, men er ikke begrænset til at:
a) betjene, ændre tilstanden af eller styre udstyret enten gennem en grænseflade (fx software eller hardware) eller gennem betjeneren af udstyret, eller
b) levere output relateret til (hardware) funktionen af dette udstyr.
Note: Software, der styrer eller påvirker brugen af (hardware) medicinsk udstyr, kan være klassificeret som et tilbehør til et (hardware) medicinsk udstyr.
Medicinsk udstyr-software er software, hvis erklærede formål er at blive anvendt alene eller i kombination, til et formål som beskrevet i definitionen af medicinsk udstyr14 eller et medicinsk udstyr til in vitro-diagnostik15.
State-of-the-art
Det nuværende niveau af teknisk kunnen og/eller accepteret klinisk praksis i forhold til produkter, processer og patienthåndtering, baseret på det relevante underbyggede fund i videnskaben, teknologi og erfaring.
Software skal i sig selv have et medicinsk formål for at blive kvalificeret som medicinsk udstyr-software (MDSW). Det skal bemærkes, at det erklærede formål, som beskrevet af fabrikanten af softwaren, er relevant for kvalificeringen og klassificeringen af udstyret.
For at blive kvalificeret som medicinsk udstyr-software, skal produktet først opfylde definitionen af software, som nævnt i denne vejledning, og definitionen af et medicinsk udstyr. For at blive kvalificeret som et medicinsk udstyr til in vitro-diagnostik software, skal produktet dertil opfylde definitionen af medicinsk udstyr til in vitro-diagnostik.
Hvor et givent produkt ikke er omfattet af definitionen for medicinsk udstyr, eller er ekskluderet af anvendelsesområdet af forordningerne om medicinsk udstyr, kan der dog være anden lovgivning, som finder anvendelse. Software, som ikke er omfattet af definitionen af medicinsk udstyr eller medicinsk udstyr til in vitro-diagnostik (software der ikke er MDSW), men hvis erklærede formål fra fabrikanten er at være et tilbehør til medicinsk udstyr eller medicinsk udstyr til in vitro-diagnostik, vil falde under den respektive forordning enten Forordning (EU) 2017/745 – MDR eller Forordning (EU) 2017/746 – IVDR.
Eksempler på software i en sundhedsmæssig sammenhæng kan være, software der direkte kan styre et (hardware) medicinsk udstyr (fx strålebehandlingssoftware), software der kan give øjeblikkelig beslutningstræffende information (fx blodsukkermålersoftware) eller software der kan give støtte til sundhedspersonale (fx EKG-fortolkningssoftware).
Det er vigtigt at klarlægge, at ikke al software anvendt indenfor sundhedspleje er kvalificeret som medicinsk udstyr. For eksempel, ”Simpel søgning”, som refererer til udtræk af journaler ved at sammenligne journal metadata med journalsøgekriterier eller udtræk af information, er ikke kvalificeret som medicinsk udstyr (fx opslagsfunktioner).
Dog kan software, hvis formål er at behandle, analysere, skabe eller modificere medicinsk information, kvalificeres som medicinsk udstyr, hvis skabelsen eller modifikationen af denne information er styret af et medicinsk erklæret formål. For eksempel vil software, som ændrer fremstillingen af data til et medicinsk formål, kvalificere sig som medicinsk udstyr-software. Dette kan være ”at søge efter et billede som understøtter en klinisk hypotese i sammenhæng med en diagnose eller behandlingsudsigt” eller ”software der lokalt forstærker kontrasten af fund i en billedfremstilling, så det kan understøtte beslutningsstøtte eller handlinger udført af brugeren”.
Software hvis formål er ikke-medicinske (udover MDR Bilag XVI udstyr, produkter uden et medicinsk formål), såsom fakturering eller personaleplanlægning, kvalificerer sig ikke som medicinsk udstyr-software.
En opgave såsom at sende e-mail, internet- eller stemmebeskeder, dataparsing, tekstbehandling og backup bliver i sig selv ikke anset som et medicinsk formål.
Dertil kan software afvikles på forskellige styresystemer eller i virtuelle miljøer. Disse styresystemer eller virtuelle miljøer har ikke indflydelse på kvalifikationskriteriet.
Det skal fremhæves, at risikoen for skade på patienter, brugere af softwaren, eller enhver anden person, relateret til brugen af softwaren indenfor sundhedspleje, herunder en mulig fejl, ikke er et kriterie for, om softwaren kvalificerer sig som medicinsk udstyr.
Medicinsk udstyr-software er software, som er tilsigtet at blive anvendt alene eller i kombination , med et formål som er omfattet af definitionen af et ”medicinsk udstyr” i MDR eller IDVR, uanset om softwaren er uafhængig eller styrer eller påvirker brugen af medicinsk udstyr.
Note 1: MDSW kan være uafhængigt ved at have sit eget medicinske formål og dermed møde definitionen af et medicinsk udstyr eller medicinsk udstyr til in vitro-diagnostik i sig selv. Dette gør sig eksempelvis gældende i følgende tilfælde:
MDSW der anvender moderens parametre såsom alder, koncentration af serummarkører og information opnået gennem føtale ultralydsundersøgelse til evaluering af Downs syndrom (trisomi 21).
MDSW der modtager målinger fra transrektale ultralydsfund, alder, og in vitro-diagnostiske instrumenter og beregner en patients risiko for at udvikle prostatacancer.
MDSW-smartwatch app, hvis tilsigtede brug er at sende alarmnotifikationer til brugeren og/eller sundhedsperson, når denne genkender uregelmæssig hjerterytme med detektion af hjertearytmi som formålet.
Note 2: Hvis softwaren styrer eller påvirker brugen af (hardware) medicinsk udstyr, og som også har et medicinsk formål, er dette klassificeret som medicinsk udstyr. Dette gør sig eksempelvis gældende i følgende tilfælde:
Billedanalysesoftware til analyse af melanomer hvis tilsigtede brug er at styre en næsten-infrarød laserscanner.
MDSW hvis tilsigtede brug er at måle og sende blodsukkerniveauer, udregne insulindosiskrav og styre insulinpumpen så denne giver den beregnede dosis (closed loop insulin infusionssystem).
Note 3: Software kan være kvalificeret som MDSW, uafhængigt af dennes fysiske lokation (fx hvis denne er placeret i en cloud, på en computer, på en mobiltelefon eller som en ekstrafunktion i et andet hardware medicinsk udstyr). Dette gør sig eksempelvis gældende i følgende tilfælde:
MDSW hvis tilsigtede brug er at fjernstyre en point-of-care-testning/patientnær testning fra en fjerntliggende lokation.
Note 4: Et MDSW kan være tilsigtet anvendt af en sundhedsperson eller en lægmand (fx patienter eller andre personer)1617. Dette gør sig eksempelvis gældende i følgende tilfælde:
MDSW som giver insulindosisanbefalinger til en patient_, uafhængigt af leveringsmetoden af den ordinerede dosis, der kan være enten via en insulinpumpe, insulinpen eller insulinsprøjte._
Fabrikanter skal sikre, at alle regulatoriske krav som omhandler at gøre tilgængelig på markedet og overensstemmelsesvurdering er blevet overholdt. Ifølge artikel 7 i MDR og IVDR, indebærer dette også anprisninger, relateret til det medicinske formål for MDSW, som skal være understøttet af klinisk evidens. Hvis dette ikke er tilfældet, vil softwaren ikke møde forordningernes krav og må på den baggrund ikke CE-mærkes som medicinsk udstyr eller fremsætte disse anprisninger.
Software, der styrer eller påvirker brugen af et (hardware) medicinsk udstyr, hvor softwaren i sig selv ikke har et medicinsk formål og ikke i sig selv genererer information for et eller flere medicinske formål, som beskrevet i definitionen af et medicinsk udstyr eller medicinsk udstyr til in vitro-diagnostik.
Denne software kan, men er ikke begrænset til at:
a) betjene, påvirke tilstanden af eller kontrollere udstyret enten gennem en grænseflade (fx software eller hardware) eller via betjeneren af dette udstyr
b) eller forsyne output relateret til (hardware) funktionen af dette udstyr.
Note: Software, der styrer eller påvirker brugen af medicinsk udstyr, er omfattet af forordningerne for medicinsk udstyr enten som en del af/komponent af et udstyr eller som et tilbehør til et medicinsk udstyr. (se Figur 1, boks 2). Dette gør sig eksempelvis gældende i følgende tilfælde:
Software hvis tilsigtede brug er at betjene et klinisk kemisk analyseudstyr.
Software med indbygget elektroniske kontroller til IVD-kvalitetskontrolprocedurer. Disse kvalitetskontrolprocedurer skal give brugere en forsikring af, at udstyret fungerer efter specifikationerne.
Beslutningstrin 1: hvis produktet er software ifølge kapitel 1 (Definitioner og forkortelser) i denne vejledning, kan dette være medicinsk udstyr-software, fortsæt til beslutningstrin 2. Hvis produktet ikke er software ifølge definitionen i denne vejledning, er dette ikke dækket af denne vejledning, men kan stadigvæk være dækket af forordningerne om medicinsk udstyr.
Beslutningstrin 2: hvis produktet er et MDR Bilag XVI udstyr (produkt uden et medicinsk formål), eller er tilbehør til et medicinsk udstyr18, eller er et software, der styrer eller påvirker brugen af et medicinsk udstyr, må det betragtes som en del af det udstyr i dettes regulatoriske proces eller uafhængigt, hvis det er et tilbehør. Hvis dette er tilfældet, er produktet dækket af forordningerne for medicinsk udstyr. Hvis dette ikke er tilfældet, fortsæt til beslutningstrin 3.
Beslutningstrin 3: hvis softwaren udfører en handling på data, eller udfører en handling udover lagring, arkivering, kommunikation19, simpel søgning, tabsfri kompression (fx ved brug af en kompressionsprocedure der tillader eksakt rekonstruktion af den originale data) kan dette være et medicinsk udstyr-software (Se afsnit 2.1 for mere vejledning omkring disse softwarefunktioner). Hvis dette er tilfældet, fortsæt til beslutningstrin 4. Hvis dette ikke er tilfældet, er produktet ikke dækket af forordningerne for medicinsk udstyr.
Beslutningstrin 4: er handlingen til gavn for individuelle patienter?
Eksempler, hvor software ikke er betragtet som værende til gavn for individuelle patienter, er dem, som er tiltænkt til kun at samle befolkningsdata, give en generisk diagnose eller behandlingsveje (ikke målrettet individuelle patienter), videnskabelig litteratur, medicinske atlas, modeller eller skabeloner såvel som software, som kun er tiltænkt til brug i epidemiologiske studier eller registre. Er handlingen til gavn for individuelle patienter, fortsæt til beslutningstrin 5. Hvis dette ikke er tilfældet, er produktet ikke dækket af forordningerne for medicinsk udstyr.
Beslutningstrin 5: er softwaren et medicinsk udstyr-software (MDSW) ifølge definitionen i denne vejledning? Hvis dette er tilfældet, er produktet dækket af forordningerne for medicinsk udstyr. Hvis dette ikke er tilfældet, er produktet ikke dækket af forordningerne for medicinsk udstyr.
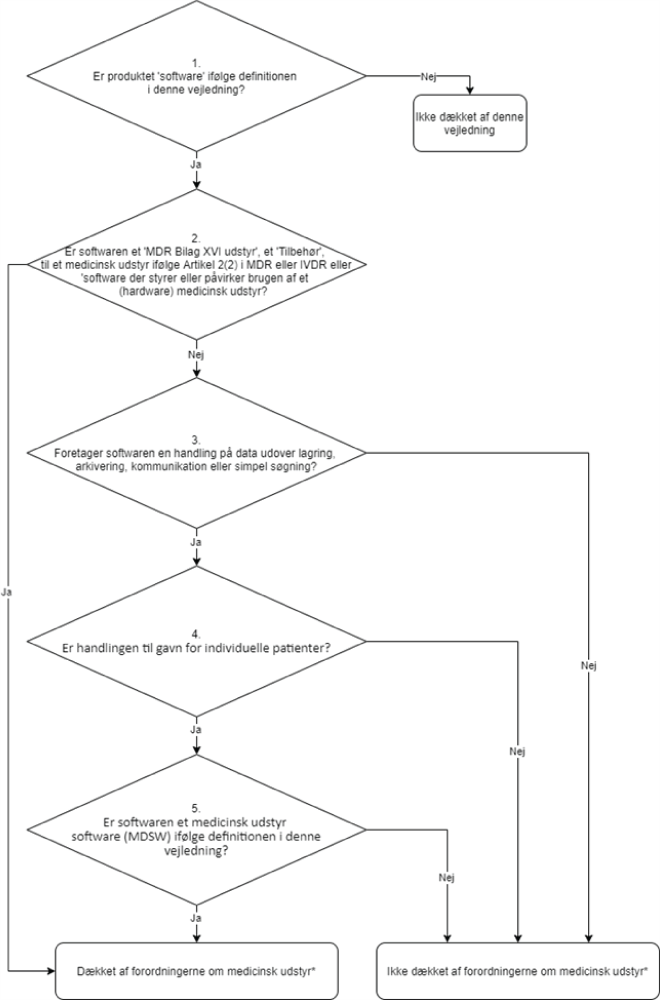
Figur 1 Beslutningstrin til hjælp med at afgøre om et produkt kan kvalificeres som et MDSW
Forordninger om medicinsk udstyr* refererer til forordninger: Forordning (EU) 2017/745 – MDR og Forordning (EU) 2017/746 – IVDR.
Medicinsk udstyr-software (MDSW), der opfylder definitionen af medicinsk udstyr til in vitro-diagnostik, er omfattet af Forordning (EU) 2017/746 om in vitro-diagnostisk medicinsk udstyr. Såfremt fabrikanten specifikt har tiltænkt, at MDSW skal anvendes sammen med et medicinsk udstyr til in vitro-diagnostik, for at muliggøre, at dette anvendes i overensstemmelse med dettes erklærede formål, falder dette MDSW under Forordningen om in vitro-diagnostisk medicinsk udstyr, og det skal behandles som et in vitro-diagnostisk medicinsk udstyr MDSW (IVD MDSW) i sig selv.
I tilfælde hvor software styrer eller påvirker brugen af (hardware) medicinsk udstyr, bør softwaren betragtes som omfattet af den respektive forordning for det (hardware) medicinske udstyr, der styres eller hvis brug påvirkes.
Det kan f.eks. være software, der analyserer og fortolker den optiske densitet (OD) fra output fra en ELISA-reader, eller foretager analyse og fortolkning af mønster efter en blotning (linjer el prikker). En sådan software analyserer rådata outputs og anvender kliniske algoritmer til diagnostiske og/eller prognostiske formål – dermed vil softwaren kvalificeres som IVD MDSW.
Beslutningstrin 1: Tilvejebringer det medicinske udstyr software (MDSW) information indenfor definitionen af medicinsk udstyr til in vitro-diagnostik?
MDSW som tilvejebringer information ifølge Forordning (EU) 2017/746 – IVDR, artikel 2, stk. 2, litra a til f bør kvalificeres som in vitro-diagnostisk medicinsk udstyr-software (IVD MDSW)
a) om en fysiologisk eller patologisk proces eller tilstand
b) om medfødte fysiske eller mentale handicap
c) om disposition for en medicinsk tilstand eller sygdom
d) til fastlæggelse af sikkerhed for og kompatibilitet med potentielle recipienter
e) til forudsigelse af reaktioner på behandlinger
f) til definition eller monitorering af terapeutiske foranstaltninger.
Et MDSW som omfattet af definitionen i Forordning (EU) 2017/745 - MDR bør kvalificeres som Medicinsk Udstyr-Software (MD MDSW). Helt specifikt bør det overvejes, hvorvidt informationen, som softwaren tilvejebringer, understøtter følgende funktioner:
a) diagnosticering, forebyggelse, monitorering, forudsigelse, prognose, behandling eller lindring af sygdomme
b) diagnosticering, monitorering, behandling, afhjælpning af eller kompensation for skader eller handicap
c) afprøvning, udskiftning eller ændring af anatomien eller en fysiologisk eller patologisk proces eller tilstand
d) udstyr til svangerskabsforebyggelse eller -støtte
e) produkter, der specifikt er beregnet til rengøring, desinfektion eller sterilisering af udstyr som omhandlet i artikel 1, stk. 4, og Bilag XVI produkter (produkter uden et medicinsk formål).
Hvis det medicinske udstyr software (MDSW) tilvejebringer information indenfor definitionen af medicinsk udstyr til in vitro-diagnostik, fortsæt til beslutningstrin 2. Hvis det medicinske udstyr software (MDSW) ikke tilvejebringer information indenfor definitionen af medicinsk udstyr til in vitro-diagnostik er softwaren dækker af Forordning (EU) 2017/745 – MDR.
Beslutningstrin 2: Skaber dette MDSW information baseret på data udelukkende tilvejebragt af medicinsk udstyr til in vitro-diagnostik?
Hvis informationen, der er genereret, er baseret på data udelukkende tilvejebragt af medicinsk udstyr til in vitro-diagnostik, er dette software et medicinsk udstyr til in vitro-diagnostik og dermed et IVD MDSW.
Hvis den analyserede data er tilvejebragt gennem en kombination af både medicinsk udstyr til in vitro-diagnostik og medicinsk udstyr, fortsæt til beslutningstrin 3.
Beslutningstrin 3: Er det erklærede formål væsentligt drevet af datakilder, der kommer fra medicinsk udstyr til in vitro-diagnostik? Hvis ja, er den gældende lovgivning Forordning (EU) 2017/746. Hvis det erklærede formål er væsentligt drevet af datakilder, der kommer fra medicinsk udstyr, er den gældende lovgivning Forordning (EU) 2017/745.
I tilfældet hvor det erklærede formål af dette MDSW-output data opfylder definitionerne for både medicinsk udstyr og medicinsk udstyr til in vitro-diagnostik ifølge MDR og IVDR (se beslutningstrin 2), bør en vægtning af datakilder baseret på vigtigheden af denne information20 i forhold til at opfylde det erklærede formål blive gennemført for at hjælpe fabrikanten med at beslutte hvilken forordning, der finder anvendelse.
Kapitel 10 i denne vejledning tilbyder nogle eksempler på, hvordan en sådan vægtning kan blive udført.
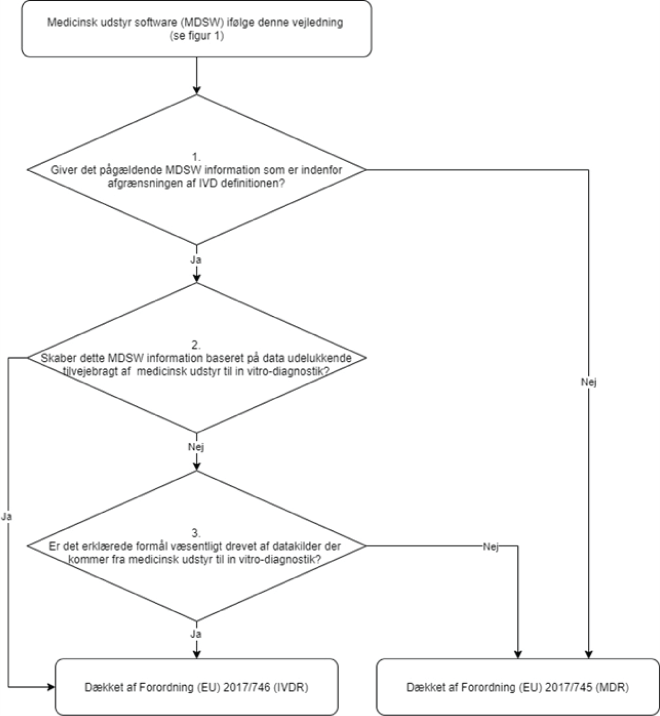
Figur 2 Beslutningstrin til hjælp med kvalificering af MDSW som enten et in vitro-diagnostisk medicinsk udstyr-software (IVD MDSW) eller som et medicinsk udstyr-software (MD MDSW)
For at CE-mærke medicinsk udstyr, skal udstyret være omfattet af definitionen for medicinsk udstyr (kvalificeres) og klassificeres korrekt i overensstemmelse med klassificeringsreglerne i bilag VIII til forordning (EU) 2017/745. Medicinsk udstyr er delt ind i fire risikoklasser, I, IIa, IIb og III. Klasse I er forbundet med den laveste risiko, mens klasse III er forbundet med den højeste risiko. Ved klasse I udstyr står fabrikanten selv for processen omkring CE-mærkningen. For medicinsk udstyr i højere risikoklasser, skal der benyttes et bemyndiget organ til at certificere udstyret.
Alle gennemførelsesbestemmelser i bilag VIII, kapitel II til forordning (EU) 2017/745 bør overvejes i forbindelse med klassificering af MDSW. Man bør specielt overveje gennemførelsesbestemmelser 3.3 og 3.5 ved klassificering af software som medicinsk udstyr.
Særlige overvejelser vedrørende gennemførelsesbestemmelse 3.3 og 3.5:
Første sætning i gennemførelsesbestemmelse 3.3 i bilag VIII præciserer reglerne der gælder for software, som influerer brugen af et medicinsk udstyr:
'Software, som styrer udstyr eller påvirker anvendelsen af udstyr, henhører under samme klasse som det pågældende udstyr. '
Anden sætning i gennemførelsesbestemmelse 3.3 i bilag VIII præciserer reglerne der gælder for uafhængig MDSW:
'Hvis softwaren er uafhængig af andet udstyr, klassificeres den selvstændigt. '
Det betyder, at software som medicinsk udstyr, der både opnår sit eget erklærede formål og også styrer eller påvirker brugen af et (hardware) udstyr til et medicinsk formål er klassificeret alene baseret på det erklærede formål, der er opnået. I et sådant tilfælde kan risikoklassen ikke være lavere end risikoklassen for det hardware medicinske udstyr.
Gennemførelsesbestemmelse 3.5 i bilag VIII er relevant for alt medicinsk udstyr-software, her fremgår det, at
‘Hvis flere regler, eller hvis flere underregler inden for samme regel, finder anvendelse på samme udstyr på grundlag af udstyrets erklærede formål, er det den strengeste regel og underregel, der medfører den højeste klassificering, der finder anvendelse. '
Eksempel på gennemførelsesbestemmelse 3.5:
Melanom-billedanalysesoftware tilsigtet til brug med en nær-infrarød laserlysscanner, der betragtes som klasse IIa i henhold til klassificeringsregel 10. Softwaren “styrer eller påvirker brugen af” den nær-infrarøde laserlysscanner, da den er beregnet til at tage kontrol over scanneren ved at lade den udføre proprietære multieksponeringsprogrammer til påvisning af melanom. Umiddelbart vil gennemførselsbestemmelse 3.3 være den gældende. Regel 11 vil dog også gælde baseret på det erklærede medicinske formål, som softwaren har. Det vil sige kræftdiagnose. Melanom-billedanalysesoftware vil derfor i stedet klassificeres som klasse III baseret på regel 11 (se afsnit 3.2 om Klassificeringsregler), og jf. gennemførelsesregel 3.5 i bilag VIII.
Klassificeringsreglerne for medicinsk udstyr herunder software er beskrevet i bilag VIII kapitel 3 (MDR). I det følgende vil der være en gennemgang og fortolkning af de væsentligste klassificeringsregler i forbindelse med klassificering af MDSW.
Klassificeringsregel 11 blev introduceret i MDR og er beregnet til at adressere de risici, der er forbundet med de oplysninger, der leveres af et aktivt udstyr, for eksempel MDSW. Regel 11 beskriver især, og kategoriserer, betydningen af de leverede oplysninger fra det aktive udstyr til patienthåndteringen i kombination med sundhedssituationen (patientens tilstand). Reglen dækker det meste medicinsk udstyr-software, og er derfor essentiel, når man klassificerer sit udstyr.
Software defineres som aktivt udstyr, hvilket betyder at klassificering af aktive (hardware) udstyr, som også indeholder MDSW, og giver information til patienthåndtering, bør overveje klassificeringsreglerne 9, 10, 12, 13, 15 og 22 i bilag VIII i MDR. Det er den strengeste regel eller underregel, der bør gælde jf. gennemførelsesbestemmelsen 3.5.
Alle de nævnte klassificeringsregler er beskrevet yderligere herunder.
Regel 11 siger:
Software, der er beregnet til at tilvejebringe oplysninger, der anvendes til at træffe beslutninger med diagnostiske eller terapeutiske formål, er klassificeret som klasse IIa, medmindre sådanne beslutninger har en virkning, der kan forårsage:
— dødsfald eller en uoprettelig forringelse af en persons sundhedstilstand, idet det i så fald henhører under klasse III, eller
— en alvorlig forringelse af en persons sundhedstilstand eller et kirurgisk indgreb, idet det i så fald er klassificeret som klasse IIb.
Software, der er beregnet til at monitorere fysiologiske processer, er klassificeret som klasse IIa, medmindre det er beregnet til at monitorere vitale fysiologiske parametre, hvor variationer af disse parametre har en sådan karakter, at de kan udgøre en umiddelbar fare for patienten, idet det i så fald er klassificeret som klasse IIb.
Alt andet software er klassificeret som klasse I.
Teksten i regel 11 kan opdeles i følgende tre underregler:
• 11a: (3 første afsnit i regel 11) beregnet til at give information, som bruges til at tage beslutninger med diagnostiske eller terapeutiske formål.
• 11b: (afsnit 4 i regel 11) beregnet til at monitorere fysiologiske processer eller parametre.
• 11c: (afsnit 5 i regel 11) alle andre anvendelser.
Underregel 11a):
Formuleringen “beregnet til at tilvejebringe oplysninger, der anvendes til at træffe beslutninger med diagnostiske eller terapeutiske formål ” beskriver i meget generelle vendinger den “handlingsmåde”, som er karakteristisk for MDSW. Derfor er denne underregel generelt gældende for MDSW, med undtagelse af MDSW, der ikke har noget medicinsk formål.
Underregel 11a) angiver, at MDSW (beregnet til at tilvejebringe oplysninger, der anvendes til at træffe beslutninger med diagnostiske eller terapeutiske formål) klassificeres som klasse IIa.
Der er to undtagelser til underregel 11a), som har til formål at anvende en strengere risikoklassifikation. Undtagelserne tager afsæt i betydningen af de leverede oplysninger, og derigennem den potentielle konsekvens af en (forkert) beslutning, truffet ved hjælp af oplysninger fra MDSW, har for brugeren. Det vil sige, at et MDSW, der er beregnet til at tilvejebringe information, der bruges til at træffe beslutninger med diagnose og terapeutiske formål, vil være i en højere risikoklasse, hvis beslutningerne der tages på baggrund af forkerte oplysninger fra MDSW kan forårsage:
i. dødsfald eller en uoprettelig forringelse af en persons sundhedstilstand, idet det i så fald henhører under klasse III, eller
ii. en alvorlig forringelse af en persons sundhedstilstand eller et kirurgisk indgreb, idet det i så fald er klassificeret som klasse IIb.
MDR indeholder flere henvisninger til "alvorlig forringelse af en persons sundhedstilstand" og “Kirurgisk indgreb”, især i sammenhæng med markedsovervågning eller klinisk undersøgelse.
Underregel 11b):
MDSW, der er beregnet til at overvåge fysiologiske processer, vil under de fleste omstændigheder ”tilvejebringe oplysninger, der anvendes til at træffe beslutninger med diagnostiske eller terapeutiske formål” og dermed falde under underregel 11a. Underregel 11b) bør derfor betragtes som en specifik regel for MDSW, der kun er gældende i tilfælde, hvor MDSW har en overvågningsfunktion. Underregel 11b) blev indført for at sikre, at MDSW, der har det samme erklærede formål som (hardware) udstyr, der falder ind under regel 10, tredje led, er i samme risikoklasse.
Denne underregel gælder dog for MDSW beregnet til brug for overvågning af alle typer af fysiologiske processer, og ikke kun vitale fysiologiske processer (svarende til regel 10, tredje led).
Vitale fysiologiske processer og parametre er fx respiration, puls, cerebrale funktioner, blodgasser, blodtryk og kropstemperatur.
Underregel 11c):
Underregel 11c) indebærer, at al anden MDSW er klassificeret som risikoklasse I.
Da softwareudstyr ikke fysisk kan administrere og/eller fjerne stoffer, henvises der i stedet til gennemførelsesbestemmelse 3.3 i bilag VIII for MDSW, der er omfattet af denne regel. Læs mere i afsnit 3.1.
Hvis et aktivt udstyr ikke er dækket af andre klassificeringsregler om aktivt udstyr (regel 9 til 12), er det klassificeret som udstyr i risikoklasse I.
Regel 15 gælder for udstyr, der anvendes til prævention eller forebyggelse af transmission af seksuelt overførte sygdomme. Software, der anvendes til prævention, klassificeres som klasse IIb.
Terapeutisk aktivt udstyr med en integreret eller inkorporeret diagnostisk funktion, som i betydelig grad er bestemmende for udstyrets patientbehandling, såsom lukkede kredsløbssystemer eller automatiske eksterne defibrillatorer, er klassificeret som klasse III.
Ligesom for medicinsk udstyr, skal software som in vitro-diagnostisk medicinsk udstyr, for at kunne CE-mærkes, være omfattet af definitionen af medicinsk udstyr til in vitro-diagnostik (kvalificeres) og klassificeres korrekt i overensstemmelse med klassificeringsreglerne i bilag VIII til Forordning (EU) 2017/746 - IVDR. IVD-udstyr er delt ind i fire risikoklasser, A, B, C og D. Klasse A er forbundet med den laveste risiko, mens klasse D er forbundet med den højeste risiko. Ved klasse A udstyr står fabrikanten selv for processen omkring CE-mærkningen. For medicinsk udstyr i højere risikoklasser, skal der benyttes et bemyndiget organ til at certificere udstyret.
Alle gennemførelsesregler i bilag VIII til Forordning (EU) 2017/746 - IVDR bør overvejes i forbindelse med kategorisering af IVD MDSW.
Særlige overvejelser om gennemførelsesregler 1.4 og 1.9:
Gennemførelsesregel 1.4 gælder kun for software, der styrer eller påvirker brugen af et in vitro-diagnostisk medicinsk udstyr. Denne regel bør som minimum overvejes som en orientering til at finde den rette (minimums) klassificering af software, der markedsføres i kombination med et (hardware) medicinsk udstyr til in vitro-diagnostik.
I henhold til anden sætning i gennemførelsesregel 1.4, hvis softwaren er uafhængig af andet medicinsk udstyr til in vitro-diagnostik, skal det klassificeres i sig selv.
Eksempler på anvendelse af denne gennemførelsesregel i henhold til forordningen om medicinsk udstyr til in vitro-diagnostik:
– Software, der udelukkende er beregnet til at styre eller påvirke brugen af et instrument beregnet af fabrikanten, der specifikt skal anvendes til in vitro-diagnostiske procedurer, er klassificeret i samme klasse som instrumentet.
Gennemførelsesregel 1.9 siger, at hvis flere klassificeringsregler gælder for det samme udstyr baseret på udstyrets erklærede formål, vil reglen, der resulterer i højere risikoklassificering, gælde.
Ved bestemmelse af den korrekte klassificering af MDSW under IVDR skal fabrikanten overveje alle klassificerings- og gennemførelsesregler i bilag VIII til Forordningen (EU) 2017/746 - IVDR.
Som beskrevet i gennemførelsesregel 1.1 i bilag VIII til Forordning (EU) 2017/746 - IVDR, er anvendelsen af klassifikationsreglerne afhængig af det erklærede formål med MDSW.
Vejledning i anvendelsen af IVD-klassificeringen og gennemførelsesregler kan findes i MDCG 2020-16 Guidance on Classification Rules for in vitro Diagnostic Medical Devices under Regulation (EU) 2017/746 på EU Kommissionens hjemmeside, se mere i afsnit 16.
Eksempler på klassificering af MDSW under IVDR:
• Software beregnet til at blive installeret i et fuldautomatisk ELISA-udstyr (enzyme-linked immunosorbent assay), og tiltænkt til at beregne den humane HbA1c-koncentration i serum ud fra resultater opnået med den pågældende HbA1c ELISA, tiltænkt screening for og diagnosticering af diabetes samt monitorering af diabetespatienter, skal være i klasse C i henhold til regel 3 (k).
• Software i et automatiseret PAP-cervikal cytologiscreeningssystem tiltænkt klassificering af PAP cervikal smear som enten normal eller suspekt, skal være i klasse C jf. regel 3 (h).
• Software til fortolkning af automatiserede aflæsninger af HIV line immunoassay - til konfirmation og bestemmelse af antistoffer mod HIV-1, HIV-1 gruppe O og HIV-2 hos mennesker i serum og plasma, skal være i klasse D i henhold til regel 1.
• Software, der bruger parametre fra moderen som alder, koncentration af serummarkører og information opnået ved føtal ultralydsundersøgelse til vurdering af risikoen for trisomi 21, skal være i klasse C i henhold til regel 3 (l).
Klassificeringseksemplerne i kapitel 11 er vejledende og har til formål at illustrere, hvordan en bestemt regel kan anvendes på et udstyr. Den angivne klassificering i eksemplet er ikke en bekræftelse af den endelige klassificering af udstyr, da andre regler også skal overvejes.
Typen af sammenkobling mellem MDSW og udstyret (fx indlejrede systemer, ledninger, Wi-Fi, Bluetooth) påvirker ikke kvalificeringen af softwaren som et udstyr under MDR og IVDR (fx om softwaren er integreret i et udstyr eller er placeret et andet sted). Dog kan MDSW være markedsført på to forskellige måder: som et medicinsk udstyr / in vitro-diagnostisk medicinsk udstyr i sig selv eller som en integreret komponent / en del af et hardware medicinsk udstyr/medicinsk udstyr til in vitro-diagnostik.
MDSW kan markedsføres eller tages i brug direkte uden at indgå som en del af andet udstyr, for eksempel:
• MDSW beregnet til at blive installeret på et fuldautomatisk enzym-bundet immunosorbent assay (ELISA) analysator og beregnet til at bestemme den humane HbA1c-koncentration i serum fra resultaterne opnået med en human HbA1c ELISA.
• MDSW-app, der giver en 10-årig risiko for hjerte-kar-sygdomme fra datainput fra en lægmand.
Overensstemmelsesvurdering:
Et MDSW markedsført som et udstyr eller taget i brug i sig selv, skal gennemgå en passende overensstemmelsesvurderingsprocedure, der tager højde for kvalifikationen, klassificeringen og det erklærede formål med MDSW.
MDSW kan markedsføres eller tages i brug som en integreret komponent eller som en del af et udstyr. Dette kan eksempelvis være ved:
• MDSW indeholdt i en blodgasanalysator, der gør det muligt for en bruger at udføre tests på et instrument.
• MDSW, der er en del af et håndholdt hardwareudstyr beregnet til patientnær testning (POCT: point-of-care-test) til bestemmelse af blodsukkerkoncentrationen.
Overensstemmelsesvurdering:
MDSW, der udelukkende markedsføres eller tages i brug som en integreret komponent / del af et (hardware) udstyr behøver muligvis ikke at gennemgå sin egen overensstemmelsesvurderingsprocedure. I dette tilfælde skal MDSW vurderes gennem den overensstemmelsesvurderingsprocedure, der anvendes på udstyret som helhed, når det placeres på markedet.
Anvendelse af klassifikationsreglerne på disse hardwareudstyr, som er en kombination af hardwareudstyr og MDSW, kræver nøje overvejelse af det erklærede formål med MDSW. Dette skal også analyseres, når der senere foretages ændringer til det pågældende MDSW.
Klinisk evaluering (MDR) / ydeevne evaluering (IVDR) er en løbende proces, udført gennem hele livscyklussen for et MDSW. Det er en struktureret, gennemsigtig, iterativ og løbende proces, som er en del af kvalitetsstyringssystemet for udstyret. Software, som kvalificerer sig som et medicinsk udstyr eller medicinsk udstyr til in vitro-diagnostik, er underlagt de samme generelle principper for klinisk evaluering (MDR) / ydeevne evaluering (IVDR), som beskrevet i gældende vejledning og regulatoriske dokumenter, som andet medicinsk udstyr eller medicinsk udstyr til in vitro-diagnostik, som fx:
• Etablere og vedligeholde en klinisk evaluering (MDR) / ydeevne evaluering (IVDR) plan og kriterier anvendt til at generere den nødvendige kliniske evidens baseret på udstyrets karakteristik.
• Identifikation af den relevante data vedrørende ydeevne og/eller sikkerhed for udstyret og alle andre resterende uadresserede problemer og mangler i data.
• Vurdering af det relevante data med hensyn til kvalitet og dettes bidrag til den kliniske evaluering (MDR) / ydeevneevaluering (IVDR).
• Analyse af den tilgængelige data og dennes relevans i forhold til at demonstrere overensstemmelse med relevante Generelle krav til sikkerhed og ydeevne (GSPR).
• Dokumentering af den relevante data, vurdering heraf og den kliniske evidens udledt deraf, i den kliniske evaluerings (MDR) / ydeevneevaluerings (IVDR) rapport.
• Opdatering af klinisk evaluering (MDR) / ydeevne evaluering (IVDR) og dennes dokumentation gennem livscyklussen for det omhandlede MDSW med data opnået gennem implementering af fabrikantens kliniske opfølgning, efter at udstyret er bragt i omsætning / fabrikantens opfølgning af ydeevne, efter at udstyret er bragt i omsætning (PMCF /PMPF) plan.
Disse metodiske principper er afbilledet i Figur 3.
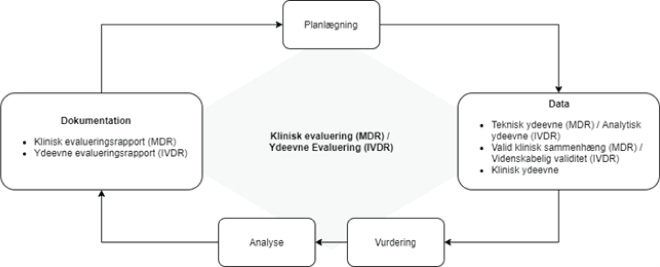
Figur 3 Overblik af stadierne i den kliniske evaluering (MDR) / ydeevne evaluering (IVDR)
Kravene for klinisk evaluering og ydeevne evaluering er skitseret i henholdsvis Artikel 61 i Forordning (EU) 2017/745 – MDR (herunder Bilag XIV) og Artikel 56 i Forordning (EU) 2017/746 – IVDR (herunder Bilag XIII).
Selvom definitionen af klinisk evaluering i Forordning (EU) 2017/745 – MDR og ydeevne evaluering i Forordning (EU) 2017/746 – IVDR ikke er identiske, er der en forventning om, at der er tilstrækkelig klinisk evidens til at demonstrere overensstemmelse med relevante generelle krav til sikkerhed og ydeevne (GSPR) under det af fabrikanten erklærede formål af udstyret. Klinisk evidens bør være tilstrækkelig og passende sammenholdt med udstyrets karakteristika, kliniske risici og dettes erklærede formål. Niveauet af kliniske evidens, der er nødvendig, skal være specificeret og begrundet af fabrikanten.
Tre hovedpunkter skal overvejes ved fremstilling af klinisk evidens for ethvert MDSW (Figur 3) og er beskrevet yderligere nedenfor.
Valid klinisk sammenhæng / Videnskabelig validitet er forstået som den grad, som outputtet fra et MDSW (fx koncept, konklusion, beregning) baseret på input og valgte algoritmer, er sammenhængende med den valgte fysiologiske tilstand eller kliniske tilstand. Denne sammenhæng skal være velbegrundet eller klinisk accepteret (fx eksistens af videnskabelige rammer eller et tilstrækkeligt niveau af evidens som uddybet i indeværende afsnit). Den valide kliniske sammenhæng / videnskabelige validitet af et MDSW skal påvises at være svarende til den kliniske situation, tilstand, indikation eller parameter, som er defineret i det erklærede formål for udstyret.
Note: Den valide kliniske sammenhæng / videnskabelige validitet forsøger at etablere fornuftige videnskabelige principper, der understøtter brugen af det omfattede MDSW. Den information som ligger til grund for etableringen af den valide kliniske sammenhæng / videnskabelige validitet skal fremsætte sammenhængen mellem det omfattede MDSW og en fysiologisk eller klinisk tilstand. Denne sammenhæng kan være kompleks at etablere. Dermed kan klinisk ydeevne fungere som ekstra information til den valide kliniske sammenhæng / videnskabelige validitet fra et klinisk synspunkt for det specifikke erklærede formål.
MDSW, som detekterer hjertearytmi ved at analysere auskultationslyde opnået gennem et digitalt stetoskop, kræver demonstrering af valid klinisk sammenhæng mellem abnorme hjertelyde og hjertearytmi.
Evidens der understøtter valid klinisk sammenhæng / videnskabelig validitet kan fx genereres gennem litteraturstudier, professionelle retningslinjer, proof of concept studier, eller fabrikantens egne kliniske undersøgelser/kliniske ydeevne studier.
Tekniske ydeevne / analytiske ydeevne valideres ved demonstrationen af evnen til nøjagtigt, pålideligt og præcist at generere det tilsigtede output, fra input data i pågældende MDSW.
Evidens der understøtter teknisk ydeevne / analytisk ydeevne kan blive genereret gennem verificerings- og valideringsaktiviteter, så som på enhedsniveau, integration, systemtest eller ved at generere ny evidens gennem brugen af anskaffede databaser, anskaffede registre, referencedatabaser eller gennem brug af tidligere indsamlet patientdata.
Klinisk ydeevne valideres ved demonstrationen af evnen til at generere et output med klinisk relevant udbytte i overensstemmelse med det erklærede formål for det pågældende MDSW. Den kliniske relevans af output fra et MDSW er en positiv indvirkning
• på helbredet på et individ, udtrykt i form af målbare, patientrelevante kliniske udfald herunder udfald relateret til diagnose, prædiktion af risiko, prædiktion af behandlingsrespons(er), eller
• relateret til MDSWs funktion, såsom screening af, monitorering, diagnose eller hjælp til diagnose af patienter, eller
• på patienthåndtering eller folkesundhed.
Evidens, som understøtter klinisk ydeevne, kan blive genereret ved at teste det pågældende MDSW under evaluering eller et tilsvarende udstyr på målgruppen og efter det erklærede formål. Den anvendte metode skal være passende til udstyrets karakteristik og erklærede formål og kan inkludere præ-klinisk testning, en klinisk afprøvning eller et klinisk ydeevnestudie.
Specifikt for MDSW, som ikke hævder at have kliniske fordele21, der kan specificeres gennem målbare, patientrelevante kliniske resultater, kan klinisk relevante outputs opnås gennem demonstreret, forudsigelig og pålidelig anvendelse og brugbarhed se i stedet afsnit 6.1 om beslutning af den valide kliniske sammenhæng / videnskabelige validitet i denne vejledning.
Dertil skal klinisk evaluering eller ydeevne evaluering af et MDSW overveje forholdet mellem fordele og risici set i lyset af state-of-the-art relaterede praksisser af lægemidler til diagnose, behandling eller patienthåndtering. Det er herudover forventet, at vurderingen af MDSW overvejer alle dele af den kliniske evaluering (MDR) / ydeevneevalueringen (IVDR) (se evt. Figur 3)
De tre punkter beskrevet ovenfor repræsenterer ikke en entydig trinvis fremgang, men viser nærmere et metodisk princip til generering af klinisk evidens.
Tilstrækkelig mængde
For at beslutte og begrunde niveauet af klinisk evidens, skal både mængden og kvaliteten af den understøttende data, evalueres. Denne vurdering kan indeholde følgende ikke-udtømmende spørgsmål:
• Understøtter data det erklærede formål, indikationer, målgrupper, kliniske påstande og kontraindikationer?
• Er de kliniske risici og den analytiske/kliniske ydeevne blevet undersøgt?
• Er karakteristika for det pågældende MDSW, såsom data input og data output, den anvendte algoritme eller typen af forbindelser blevet overvejet, når der genereres data til at understøtte udstyrets ydeevne?
• Hvad er graden af innovation/historien på markedet (hvor stor er den samlede mængde videnskabelig evidens)?
Tilstrækkelig kvalitet
• Var typen og designet af studiet/testen passende til forskningsmålet?
• Var datasættet passende og aktuelt (state-of-the-art)?
• Var den statistiske tilgang passende til at opnå en valid konklusion?
• Var alle etiske, lovmæssige og regulatoriske overvejelser/krav taget i betragtning?
• Er der nogen interessekonflikter?
Fabrikanten skal først verificere sammenhængen mellem outputtet fra det pågældende MDSW (baseret på valgte input og algoritmer) og den valgte fysiologiske/kliniske tilstand, kliniske situation eller klinisk parameter, som er defineret i det erklærede formål for det pågældende MDSW. MDSW kan indeholde flere forskellige kliniske egenskaber, styret af dettes erklærede formål, som hvert kræver en individuel vurdering.
Denne sammenhæng skal være klinisk accepteret eller velbegrundet, hvilket betyder, at den er accepteret bredt i sundhedsregi og/eller beskrevet i videnskabelig (peer-reviewed) litteratur.
Valid klinisk sammenhæng/Videnskabelig validitet kan demonstreres gennem brugen af eksisterende klinisk ydeevnedata samtidig med, at generelt anerkendt state-of-the-art også betragtes.
Valid klinisk sammenhæng/Videnskabelig validitet kan yderligere demonstreres ved at skabe ny klinisk ydeevnedata i tilfælde, hvor eksisterende data ikke er tilstrækkelig. Fx som resultat af en gap-analyse, hvor fabrikanten kunne konkludere, at der mangler yderligere data.
Eksempler på eksisterende data (i tilfældig rækkefølge):
• Tekniske standarder
• Professionelle sundhedsretningslinjer
• Systematisk videnskabelig litteraturgennemgang
• Kliniske undersøgelser/kliniske ydeevnestudier
• Publiceret klinisk data
Eksempler på at skabe ny evidens (i tilfældig rækkefølge):
• Sekundære dataanalyser (Analyse af real-world data)
• Udføre klinisk afprøvning /klinisk ydeevne studie
Fabrikanten skal verificere, at det pågældende MDSW pålideligt, præcist og konsekvent passer til det erklærede formål ved faktisk brug.
De relevante ydeevnekarakteristika, som del af GSPR’erne (generelle krav til sikkerhed og ydeevne) og som hænger sammen med de analytiske og/eller kliniske egenskaber, skal understøttes med evidens, der er genereret under verifikation og validering som del af god fremstillingspraksis (Ggood manufacturing practices) for software, eller ved at generere ny evidens gennem brugen af tilegnede databaser, tilegnede registre, referencedatabaser eller brug af tidligere indsamlet patientdata.
Teknisk ydeevne / analytisk ydeevne er bekræftet ved undersøgelsen og bestemmelsen af, at objektiv evidens for det pågældende MDSW’s specifikationer er i overensstemmelse med brugernes behov og det erklærede formål, og at kravene, der er implementeret, kan opfyldes konsekvent.
For eksempel kan ydeevneverifikation og -validering i det tilsigtede databehandlingsmiljø (hardware) og brugsmiljø karakteriseres ved demonstration af
• tilgængelighed,
• fortrolighed,
• integritet,
• pålidelighed,
• nøjagtighed (resultatet af korrekthed og præcision),
• analytisk sensitivitet,
• begrænsning for detektion,
• begrænsning for kvantificering,
• analytisk specificitet,
• linearitet,
• cut-off værdi(er),
• måleinterval (range),
• generaliseringsevne,
• forventet datahastighed eller kvalitet,
• at sårbarheder relateret til cybersikkerhed ikke findes,
• at systemet er designet med brugerens begrænsninger taget i betragtning.
Identifikation af mangler under valideringen af den tekniske ydeevne / analytisk ydeevne kunne kræve generering af ny evidens, for fx at demonstrere generaliserbarhed med virkelighedsbaserede (real-life) datasæt eller øge brugbarhedsevalueringen til tidligere ekskluderede brugergrupper.
For validering af klinisk ydeevne for et MDSW, skal fabrikanten demonstrere, at det pågældende MDSW er blevet testet efter den tilsigtede brug, målgruppe(r), brugsforhold, betjening- og brugsmiljø(er) og med alle tilsigtede brugergrupper. Afsnit 6.1 i denne vejledning giver yderligere beskrivelse af, at validering af klinisk ydeevne indeholder vurdering af klinisk sikkerhed, effektivitet, ydeevne og kan understøtte demonstrationen af kliniske fordele. Validering af den kliniske ydeevne bør overvejes for enhver ændring af softwaren til en ny opdatering. Hvis ingen validering finder sted, bør en begrundelse for dette angives i den tekniske dokumentation.
Med en validering af den kliniske ydeevne er det demonstreret, at brugere kan opnå klinisk relevante outputs gennem forudsigelig og pålidelig brug af det pågældende MDSW.
Fabrikanten bør overveje de(t) erklærede formål, indikation(er) og ønsket klinisk output angivet som anprisninger, som fører til forventede kliniske fordele som en del af valideringen af den kliniske ydeevne.
Et MDSW kan have flere egenskaber, hvoraf kun nogle af egenskaberne påstås at have kliniske fordele. Klinisk ydeevne er kun gældende for disse egenskaber. Siden MDSW kan være modulbaseret, er validering af klinisk ydeevne på modulniveau også muligt, når funktionaliteten af et modul er uafhængig af de resterende moduler. Dette vil gøre det muligt at acceptere forholdet mellem fordele og risici løbende, og kun for de MDSW-moduler, som ændrer sig. I tilfælde, hvor den endelig kombination af moduler ændrer produktets indikationer og erklærerede formål, bør ydeevnen for det endelige produkts konfiguration også evalueres. Validering af den kliniske ydeevne kan karakteriseres ved demonstration af anvendelig klinisk data til det pågældende MDSW, såsom (ikke-udtømmende):
• klinisk/diagnostisk sensitivitet,
• klinisk/diagnostisk specificitet,
• positive predictive value,
• negative predictive value,
• positive likelihood ratio,
• negative likelihood ratio,
• brugbarhed/brugergrænseflade
• konfidensinterval(ler)
Klinisk data kan opnås gennem en eller flere metoder, såsom dem der er angivet i GHTF/SG5/N7:2012 og IMDRF/SaMD WG/N41FINAL:2017.
Udover overvejelserne ovenfor, skal klinisk evaluering af klasse III og implantabelt udstyr (MDR) indeholde data fra en klinisk afprøvning medmindre, at forholdene i Artikel 61(4), (5) eller (6) i Forordning (EU) 2017/745 – MDR er opfyldt.
For MDSW, som er omfattet af Forordning (EU) 2017/746 – IVDR, kræver evalueringen af den kliniske ydeevne studier uanset udstyrets klasse, medmindre der foreligger en behørig begrundelse, der støtter sig op af andre klinisk ydeevne datakilder.
Relevante almindelige specifikationer bør også overvejes.
De praktiske og opnåelige fordele ved en klinisk afprøvning/et klinisk ydeevnestudie bør overvejes som en del af beslutningen af hvilket data, der er nødvendige for at demonstrere sikkerheden og ydeevnen for et nyt eller modificeret MDSW. Afprøvningen eller studiet bør tage højde for potentielle risici, bør følge relevante etiske krav, og bør være i overensstemmelse med alle relevante lovmæssige og regulatoriske krav.
Det bør overvejes, om det MDSW har specifikke karakteristika, når der opstilles en klinisk afprøvning eller et klinisk ydeevnestudie. Hvis det pågældende MDSW bruges til at beslutte en patients fremtidige tilstand (fx disposition, prognose, forudsigelse) eller hvis outputtet fra det pågældende MDSW påvirker et klinisk udfald (fx behandlingseffekt) eller patienthåndteringsbeslutninger, kan et prospektivt studie være nødvendigt som en del af udstyrets kliniske evaluering (MDR) / ydeevne evaluering (IVDR). I andre situationer kan et retrospektivt studie være mere relevant til at generere den nødvendige data til at sikre overholdelse af GSPR’er (generelle krav til sikkerhed og ydeevne), da der ikke er nogen indvirkning på patienthåndtering, og fordi forskningen ikke introducerer nogen risici for patienter. Sådan en tilgang er kun mulig i tilfælde, hvor der er tilstrækkelig adgang til datasæt, der indeholder en tilstrækkelig mængde data, kvalitet og som stammer fra målpopulationen.
Formelle krav i MDR Artikel 62(1), 74 og 82 skal overholdes såfremt de er relevante for præmarked retrospektive studier for MDSW, som er omfattet af MDR. Læs mere om kravene til kliniske afprøvninger i vejledning om ansøgning om tilladelse til kliniske afprøvninger.
6.6 Hvor demonstration af overensstemmelse baseret på klinisk data ikke er anset som hensigtsmæssig
På linje med bestemmelserne i MDR Artikel 61(1) og IVDR Artikel 56 (1), skal niveauet af nødvendig klinisk evidens, der kræves for et udstyr, være tilstrækkelig, når udstyrets anprisninger og karakteristika betragtes. For medicinsk udstyr, hvor demonstrationen af overensstemmelse med GSPR’er (generelle krav til sikkerhed og ydeevne) baseret på klinisk data ikke anses som værende hensigtsmæssig (MDR Artikel 61(10)), skal fabrikanten behørigt angive det i den tekniske dokumentation, hvorfor det er tilstrækkeligt at demonstrere overensstemmelse baseret udelukkende på ikke-kliniske testmetoder, herunder ydeevne evaluering, testning af udstyret (bench testing), præklinisk evaluering, og brugbarhedsvurdering.
Begrundelsen skal være baseret på udfaldet af en risikovurderingsproces. Denne bør indeholde en evaluering af klinisk state-of-the-art, herunder alternative diagnose- og behandlingsmuligheder, herunder dem der er fundet gennem litteratur, og en vurdering af deres relevans i forhold til det udstyr, som evalueres. Udstyr/krops-interaktionen, tilsigtet kliniske ydeevne, og fabrikantens anprisninger bør specifikt overvejes.
En klinisk evaluering (MDR) er stadigvæk nødvendig, og den ovenstående information og evidensbaseret begrundelse bør præsenteres i den kliniske evalueringsrapport.
Ligeledes for IVD, hvor der på grund af udstyrsspecifikke karakteristika kan være tilfælde, hvor demonstration af overensstemmelse med GSPR’er baseret på klinisk data ikke anses som hensigtsmæssigt, er en ydeevneevaluering (IVDR) stadigvæk nødvendig og en begrundelse skal være tilgængelig og dokumenteres i ydeevneevalueringsplanen og den dertilhørende ydeevne evalueringsrapport.
Fabrikanten bør udarbejde evidens, udføre en analyse af forholdet mellem fordele og risici og dokumentere den kliniske evaluering eller ydeevne evalueringen og dennes udbytte i en klinisk evalueringsrapport (MDR) / ydeevne evalueringsrapport (IVDR).
Sikkerheden, effektiviteten og ydeevnen for det pågældende MDSW bør aktivt og løbende overvåges af fabrikanten.
Data for dette kan inkludere, men er ikke afgrænset til, klager efter at udstyret er bragt i omsætning, PMCF/PMPF data, data fra ydeevne i et ikke-klinisk (real-world) miljø, direkte brugerfeedback eller ny(e) forskning/retningslinjer og bør være underlagt den kliniske evaluering (MDR) / ydeevne evaluering (IVDR) som vist i Figur 3.
Det unikke niveau af forbindelsesmuligheder for et MDSW faciliterer adgang til ikke-kliniske (real-world) ydeevne data, som kan bruges til flere formål, herunder, men ikke begrænset til:
• tidspassende detektion og rettelse af fejl.
• detektion af systematisk fejlbrug.
• forståelse af brugerinteraktioner.
• at foretage løbende monitorering af klinisk ydeevne.
• at forbedre effektiviteten.
• udvikle påstandene i den kliniske udviklingsplan (MDR) eller fremtidige udgivelser.
MDSW kan frigives med CE-mærkning med initiale anprisninger og validerede kliniske fordele. Monitorering og ikke-kliniske (real-world) ydeevne data hjælper med at opstille hypoteser til fremtidige MDSW-funktioner og erklærede formål.
Noget medicinsk udstyr-software kan blive opdelt i flere applikationer, hvor hver af disse applikationer hører sammen med et modul. Nogle af disse moduler har et medicinsk formål, andre har ikke.
Sådanne moduler kan være beregnet til at dække mange behov, fx:
• Indsamle og vedligeholde administrative patientdata.
• Arkivere patientens medicinske historie.
• Fakturering og andre bogholderiopgaver.
• Skabe en forbindelse til medicinreceptsystemer (med en mulig forbindelse til apoteker).
• Give ekspert system assistance til medicinsk beslutningsstøtte (fx strålebehandlingsdosis planlægning).
Dette rejser problemstillingen, om hvorvidt hele produktet skal være CE-mærket, når ikke alle applikationerne har et medicinsk formål.
Computerprogrammer, der anvendes i sundhedspleje, kan have applikationer, som er opbygget af både medicinsk udstyr og ikke-medicinsk udstyr moduler.
Modulerne, som er underlagt forordningen om medicinsk udstyr og forordningen om medicinsk udstyr til in vitro-diagnostik, skal overholde kravene i forordningerne om medicinsk udstyr og skal bære et CE-mærke. De ikke-medicinsk udstyr moduler er ikke underlagt kravene for medicinsk udstyr. Det er fabrikantens forpligtelse at identificere grænserne og grænseflader til de forskellige moduler.
Grænserne af modulerne, som er underlagt forordningerne om medicinsk udstyr, skal tydeligt identificeres af fabrikanten baseret på det erklærede formål.
Hvis modulerne, som er underlagt forordningerne om medicinsk udstyr, er beregnet til brug i kombination med andre moduler i den samlede softwarestruktur, andet udstyr eller hele kombinationen, herunder forbindelsessystem, skal disse være sikre og må ikke hæmme den angivne ydeevne for modulerne, som er underlagt forordningerne om medicinsk udstyr.
Fabrikanter skal evaluere de potentielle følgevirkninger, som en hvilken som helst ændring af funktioner, det erklærede formål, væsentlig design og fremstillingskarakteristikken har på softwarens kvalifikation som MDSW og dettes klassifikation (herunder klassifikationen af kombination af dette MDSW og et andet medicinsk udstyr/medicinsk udstyr til in vitro-diagnostik).
Det skal bemærkes, at en ændring eller tilføjelse af funktionalitet til et software kan medføre, at dette kvalificeres som et MDSW, eller en revurdering af klassifikationen af dette MDSW. Ligeledes kan et modul som tilføjes til et software, være et MDSW i sig selv.
Når man skal beslutte risikoklassen for en kombination af et modificeret MDSW og et medicinsk udstyr/medicinsk udstyr til in vitro-diagnostik, skal det erklærede formål og funktionaliteten af denne (nye) kombination tages i betragtning.
Hvis ændringer af certificeret udstyr kan få indflydelse på udstyrets sikkerhed og ydeevne eller de foreskrevne betingelser for anvendelsen af udstyret, skal sådanne ændringer godkendes af det bemyndigede organ.
Note: For alle MDSW skal fabrikanten garantere sikkerhed og ydeevne gennem softwarens levetid. Dette er gennem en løbende proces af klinisk og/eller ydeevne evaluering og risikostyring.
Software til medicinske formål udvikler sig hurtigt. Listen over eksempler nedenfor er ikke udtømmende. Eksemplerne er udarbejdet i lyset af state-of-the-art teknikker for at give en bedre forståelse af anvendelsen af de principper, der er beskrevet i forordningerne (MDR og IVDR). Følgende eksempler gennemgår om forskellige typer software kvalificerer sig som MDSW:
a) Hospitalsinformationssystemer
Hospitalsinformationssystemer betyder i denne sammenhæng systemer, der understøtter processen med patienthåndtering. Typisk er de beregnet til patientindlæggelse, til planlægning af patientaftaler, til forsikrings- og faktureringsformål. Disse sygehusinformationssystemer er ikke kvalificerede som medicinsk udstyr. De kan dog bruges med flere/andre moduler som beskrevet i det følgende. Disse moduler kan i sig selv være kvalificeret som medicinsk udstyr.
b) Software til beslutningssupport
Generelt er dette computerbaserede værktøjer, der kombinerer generelle medicinske informationsdatabaser og algoritmer med patientspecifikke data. De er beregnet til at give sundhedspersonale og/eller brugere anbefalinger til diagnose, prognose, overvågning og behandling af individuelle patienter.
Baseret på Figur 1 er de kvalificeret som medicinsk udstyr. Eksempler på denne type software:
• Systemer til planlægning af strålebehandling der har til formål at beregne den dosis af ioniserende bestråling, der skal påføres en bestemt patient. De anses for at kontrollere, overvåge eller direkte påvirke kilden til ioniserende stråling og er kvalificeret som medicinsk udstyr.
• Lægemiddelplanlægningssystemer (fx Kemoterapi) der har til formål at beregne den lægemiddeldosis, der skal administreres til en bestemt patient, og er derfor kvalificeret som medicinsk udstyr.
• Computerstøttede detektionssystemer der er beregnet til at give oplysninger, der kan antyde eller udelukke medicinske tilstande, er kvalificeret som medicinsk udstyr (MDSW). For eksempel vil sådanne systemer være i stand til automatisk at analysere røntgenbilleder eller fortolke EKG'er.
c) Informationssystemer
Informationssystemer, der kun er beregnet til at overføre, gemme, konvertere, formatere og/eller arkivere data er ikke kvalificerede som medicinsk udstyr i sig selv. De kan dog bruges med andre moduler, som i sig selv kan være kvalificerede som medicinsk udstyr (MDSW).
c. 1.) Elektroniske patientjournalsystemer
Elektroniske patientjournalsystemer er beregnet til at gemme og overføre elektroniske patientjournaler. De arkiverer alle slags dokumenter og data relateret til en bestemt patient. De elektroniske patientjournaler skal som udgangspunkt ikke kvalificeres som et medicinsk udstyr, idet en elektronisk patientjournal, der erstatter en patients papirjournal, ikke opfylder definitionen på et medicinsk udstyr. Modulerne, der anvendes med elektroniske patientjournalsystemmoduler, der i sig selv kan kvalificeres som medicinsk udstyr (MDSW), er for eksempel:
• En billedfremviser med funktionalitet til diagnose baseret på digitale billeder.
• Et medicinmodul
c. 1.1.) Kliniske informationssystemer - CIS / patientdatastyringssystemer - PDMS
Et CIS / PDMS er et softwarebaseret system, der primært er beregnet til at gemme og overføre patientinformation genereret i forbindelse med patientens intensivbehandling (fx på intensivafdelinger). Normalt indeholder systemet information såsom patientidentifikation, vitale intensivparametre og andre dokumenterede kliniske observationer. Disse CIS / PDMS er ikke kvalificeret som medicinsk udstyr. Moduler, der er beregnet til at give yderligere information, der bidrager til diagnose, terapi og opfølgning (fx generere alarmer), er kvalificeret som medicinsk udstyr.
c. 1.2.) EKG-system (pre-hospital electrocardiograph)
Et system til styring af præ-hospital-EKG er et softwarebaseret system beregnet til ambulancetjenester til at gemme og overføre information fra patienter til en læge på en fjerntliggende lokation. Normalt indeholder systemet information om patientidentifikation, vitale parametre og andre dokumenterede kliniske observationer. Disse præ-hospital elektrokardiografi (EKG) systemer er ikke kvalificeret som medicinsk udstyr.
Moduler, der opretter og giver ny patientbehandlingsinformation til paramedicinere eller til lægen på et fjerntliggende lokation for at starte patientens behandling, mens patienten transporteres, er kvalificeret som medicinsk udstyr.
d) Kommunikationssystemer
Sundhedssektoren bruger kommunikationssystemer (fx e-mail-systemer, mobile telekommunikationssystemer, videokommunikationssystemer, personsøgning osv.) for at overføre elektronisk information. Der sendes forskellige typer meddelelser, såsom recept, henvisninger, billeder, patientjournaler osv. De fleste kommunikationssystemer håndterer andre typer meddelelser end medicinsk information. Dette kommunikationssystem er beregnet til generelle formål og bruges til overførsel af både medicinsk og ikke-medicinsk information. Kommunikationssystemer er normalt baseret på software til generelle formål og falder ikke inden for definitionen af et medicinsk udstyr. Kommunikationssystemmoduler kan bruges sammen med andre moduler, der i sig selv kan kvalificeres som medicinsk udstyr (MDSW).
Eksempelvis er et softwaremodul, der genererer alarmer baseret på overvågning og analyse af patientspecifikke fysiologiske parametre, kvalificeret som et medicinsk udstyr (MDSW).
d.1) Telemedicinsk system
Telemedicinsk system er beregnet til at muliggøre monitorering og/eller levering af sundhedspleje til patienter fjernt fra sundhedspersonalets fysiske lokation eller få patienter til at indbringe oplysninger, der er beslutningsunderstøttende for videre diagnose eller behandling. Telemedicinske systemer, som fungerer, som kommunikation uden at have et erklæret medicinsk formål, betragtes som udgangspunkt ikke som et medicinsk udstyr.
d.1.1.) Patientrapporterede oplysninger
I sundhedssektoren bruger man i stigende grad softwareprogrammer til at få patienter til at indberette eller videregive oplysninger om deres sundhedstilstand. Patientrapporterede oplysninger (PRO) dækker over oplysninger om helbred og livskvalitet, der kommer direkte fra patienterne selv – typisk via digitale spørgeskemaer, som patienterne udfylder. Oplysninger viderebringes til sundhedspersonale, og vil herefter indgå som beslutningsunderstøttende information i patients videre udredning, diagnosticering eller behandling. Softwaren kvalificerer sig som MDSW.
Det samme gør sig gældende for softwarebaserede beslutningstræer eller algoritmer, hvor patienterne indtaster sundhedsrelaterede oplysninger, og en softwarebaseret løsning giver patienten information om eventuelt videre behandling, udredning eller diagnosticering på baggrund af disse oplysninger.
e) Websystemer til overvågning af data
Et websystem til overvågning af kliniske data interagerer typisk med et medicinsk udstyr (fx implanteret udstyr eller udstyr til hjemmepleje) og bruger en sender til at sende informationen over internettet, en fastnettelefon eller et mobilnetværk.
Oplysningerne indsamles og lagres på en webserver, som normalt drives af en ekstern part, der normalt er producenten af systemet. Oplysningerne kan nås af autoriserede sundhedspersonale eller patienten via en internetforbindelse.
• Overvågning af ydeevne for medicinsk udstyr:
Moduler, der er beregnet til at overvåge medicinsk udstyrs medicinske ydeevne, er omfattet af reglerne om medicinsk udstyr. Dette inkluderer den kliniske ydeevne og svigt, der kan påvirke udstyrets medicinske ydeevne. Et eksempel på et sådant produkt er et websystem til overvågning af aktive implantater såsom pacemakere eller Intra Cardiac Defibrillators (ICD'er).
• Overvågning af medicinsk udstyrs ikke-medicinske ydeevne
Moduler, der er beregnet til administrativ overvågning af medicinsk udstyrs ikke-medicinske ydeevne, falder ikke nødvendigvis inden for anvendelsesområdet for medicinsk udstyr, for eksempel:
Software til overvågning af medicinsk udstyr i hospitalssystemer med henblik på vedligeholdelse og reparation.
f) In vitro-diagnostisk software til medicinsk udstyr
f. 1.) Laboratorieinformationssystemer (LIS)
Laboratorieinformationssystem (LIS) er i denne sammenhæng et system, der understøtter processen fra patientprøve til patientresultat. Typisk har de præ-analytiske funktioner til bestilling, sortering og distribution af testprøver. Hovedopgaven er styring og validering af indgående information opnået fra in vitro-diagnostiske medicinske udstyrs analysatorer, der er tilsluttet systemet, såsom kalibrering, kvalitetskontrol, produktudløb og feedback (fx gen-testning af utilstrækkelige prøver) gennem sammenkoblinger med forskellige analyseinstrumenter (teknisk og klinisk validering). Endelig tillader den postanalytiske proces kommunikation af laboratorieresultater, statistik og valgfri rapportering til eksterne databaser.
Softwaren understøtter normalt følgende funktioner:
• Bestilling af laboratorietests, prøver med etiketter og sortering.
• Teknisk og klinisk validering, forbindelse til analyseinstrumenter.
• Laboratorieresultater og rapporter på papir, fax eller elektroniske poster, der kan returneres direkte til fx bestillingsklinikkens patientjournal.
• Analytiske instrumenter kan have grænseflade med Hospital Information Systems (HIS), elektroniske patientjournalsystemer, mikrobiologiske overvågningsdatabaser og alarm-systemerosv.
Bemærk: Software beregnet til at ændre repræsentationen af tilgængeligt medicinsk udstyr til in vitro-diagnostik resultater betragtes ikke som et in vitro-diagnostisk medicinsk udstyr, fx grundlæggende aritmetiske operationer (fx gennemsnit, konvertering af enheder) og / eller afbildning af resultater i funktion af tid og/eller en sammenligning af resultaterne.
Hvis eksempelvis resultaterne er tilgængelige, læsbare og forståelige uden indblanding af softwaren, er laboratorieinformationssystemer (LIS) ikke kvalificeret som medicinsk udstyr i sig selv. De kan dog bruges sammen med yderligere moduler. Disse moduler kan i sig selv være kvalificeret som medicinsk udstyr. Dette kunne være et modul, hvis formål er at vurdere kritikaliteten/vigtigheden af de krævede tests og udføre automatisk omprioritering af ordren baseret på patientdata, og det vil derfor være kvalificeret som en MDSW.
f. 2.) Fortolkning af rådata
I det tilfælde, hvor MDSW er nødvendigt for at gengive rådata, skal dette MDSW betragtes som styrende eller påvirkende af brugen af det medicinske udstyr til in vitro-diagnostik. Ved gengivelse af rådata forstås det som læsbart for brugeren, opnået fra et medicinsk udstyr til vitro-diagnostik ved hjælp af in vitro-undersøgelse af kropsprøver. Dette gør sig gældende, når et MDSW er specifikt beregnet til at blive brugt sammen med et in vitro-diagnostiske medicinske udstyr for at gøre det muligt, at det bruges i overensstemmelse med det erklærede formål.
f. 3.) Overvågning af hjemmepleje
Software beregnet til arkivering af patientresultater eller til overførsel af resultater opnået fra in vitro-diagnostisk medicinsk udstyr, fra hjemmemiljøet til sundhedsudbyderen, er ikke et in vitro-diagnostisk medicinsk udstyr. Resultaterne er tilgængelige, læselige og forståelige af brugeren uden interventionen af softwaren.
Figur 1 - Eksempel 1:
| Beslutningstrin 1 | Er produktet ’software’ dækket af definitionen i denne vejledning? |
|---|---|
| Beslutningstrin 2 | Er software et ’MDR Bilag XVI udstyr’ (produkt uden et medicinsk formål), et ’tilbehør’ til et medicinsk udstyr ifølge artikel 2 (2) fra MDR eller IVDR eller ’software der styrer eller påvirker brugen af et (hardware) medicinsk udstyr? |
| Beslutningstrin 3 | Foretager softwaren en handling på data udover lagring arkivering, kommunikation, simpel søgning? |
| Beslutningstrin 4 | Er handlingen til gavn for individuelle patienter? |
| Beslutningstrin 5 | Er softwaren et medicinsk udstyr-software (MDSW) dækket af definitionen i denne vejledning? |
| Tabel 1: Beslutningstrin for kvalificering af MDSW (fra Figur 1) |
Et softwaremodul, der afvikles på et in vitro-diagnostisk medicinsk udstyr og sporer, hvordan laboratoriet klarer sig i realtid på vigtige operationelle målinger såsom testvolumener, leveringstider, afventende tests og kvalitetskontrol. Dets hensigt er at forbedre et laboratoriums operationer ved at levere realtidsovervågning af præstationsmålinger, der kan drive ændringsstyring og løbende forbedringsinitiativer inden for laboratoriet. Softwaren kan konfigureres, så kunderne kan vælge de mål, som de gerne vil fokusere på.
Kvalifikation: Beslutningstrin 1 afsluttes med et “ja”, da softwaren er et produkt, der bruger et sæt instruktioner (eller algoritme) til at behandle inputdata og oprette outputdata. Beslutningstrin 2 bestemmer, at softwaren ikke er et MDR Annex XVI-udstyr, og det er heller ikke et tilbehør til et medicinsk udstyr eller en software, der styrer eller påvirker brugen af et medicinsk udstyr. Beslutningstrin 3 besvares "ja", da softwaren gør mere end opbevaring, arkivering, kommunikation eller simpel søgning efter information. Beslutningstrin 4 besvares "nej", da softwaren ikke udfører denne handling til fordel for de enkelte patienter. Konklusionen er, at softwaren ikke er omfattet af definitionen for medicinsk udstyr. Dette er passende, da softwaren er beregnet til at være et laboratorieinformationssystem (LIS), der ikke betragtes som et medicinsk udstyr.
Figur 2 - Eksempel 2:
| Beslutningstrin 1 | Tilvejebringer det medicinske udstyr software (MDSW) information indenfor definitionen af medicinsk udstyr til in vitro-diagnostik? |
|---|---|
| Beslutningstrin 2 | Skaber dette MDSW information baseret på data udelukkende tilvejebragt af medicinsk udstyr til in vitro-diagnostik? |
| Beslutningstrin 3 | Er det erklærede formål væsentligt drevet af datakilder, der kommer fra medicinsk udstyr til in vitro-diagnostik? |
| Tabel 2: Beslutningstrin for kvalificering af IVD MDSW (fra Figur 2) |
MDSW beregnet til at generere en risikoscore for at udløse plejeprocesser for at hjælpe med at reducere overførsler til intensivafdelinger, genindlæggelser, bivirkninger og opholdets varighed. Risikoscore inkluderer som standard åndedrætsfrekvens, puls, blodtryk og SpO2, men brugeren kan konfigurere det til at omfatte andre parametre, herunder in vitro-diagnostiske medicinske udstyrsresultater.
Kvalifikation: I beslutningstrin 1 kan MDSW forstås at opfylde kriterierne i IVDR, artikel 2, stk. 2, litra (a), (f) og (h). Svaret er derfor ”ja”. Beslutningstrin 2 besvares med et ”nej”, da et in vitro-diagnostisk medicinsk udstyrsresultat kan medtages i beregningen. Beslutningstrin 3 styrer betydningen af den afledte information fra medicinsk udstyr, som styres af det erklærede formål, hvilket resulterer i kvalificering af softwaren som MD MDSW (da data modtaget fra det in vitro-diagnostiske medicinske udstyr ikke anses for afgørende for det samlede beregningsresultat (output) opnået af MDSW).
Figur 2 - Eksempel 3: En MDSW-algoritme beregnet til at give information om den statistiske disposition for Downs syndrom (trisomi 21) og Edwards syndrom (trisomi 18) i graviditetens første og andet trimester. MDSW analyserer inputdata fra forskellige in vitro-diagnostiske medicinske udstyrsanalyser samt ultralydsmålinger af næseknoglen eller halsfoldningen. MDSW giver klinikere/fødselslæger en risikofaktor-score for et fosters sandsynlighed for at have genetiske mutationer i første eller andet trimester af graviditeten. Risikoscoren viser, om der er behov for yderligere diagnostisk test for at bekræfte de genetiske mutationer af trisomi 21, trisomi 18.
Kvalifikation: Beslutningstrin 1 kan besvares "ja", da softwaren bærer et medicinsk formål og opfylder definitionen af MDSW. MDSW opfylder kriterierne i IVDR, artikel 2, stk. 2, litra (c), da den giver information i henhold til definitionen af medicinsk udstyr til in vitro-diagnostisk brug. Beslutningstrin 2 besvares "nej", da en billeddannelsesmåling er inkluderet i beregningen. Beslutningstrin 3 svares "ja", da det erklærede formål hovedsageligt er styret af data fra in vitro-diagnostisk medicinsk udstyr, hvilket resulterer i kvalificering af softwaren som en IVD MDSW (da data modtaget fra in vitro-diagnostisk medicinsk udstyr (markører) anses for afgørende for det samlede beregningsresultat (output) opnået af MDSW).
Figur 2 - Eksempel 4: En bioinformatik-MDSW beregnet til digitalt at analysere Next Generation Sequencing rådata fra en patients sekventerede kræftgenomer. Det muliggør påvisning og visualisering af somatiske genomændringer (såsom substitutioner, små inserts og deletioner (indels), ændringer i genkopiantal og genomisk rearrangering) på tværs af et valgt antal gener. Derudover er det også i stand til at bestemme genomiske signaturer (såsom mikrosatellit instabilitet (MSI) og/eller tumor mutational burden (TMB)). Hvilke typer somatiske genomændringer og genomiske signaturer, der detekteres, afhænger af den valgte test. MDSW hjælper brugeren med at identificere og visualisere genomiske ændringer og er beregnet til at identificere somatiske genomændringer til støtte for diagnose- og behandlingsbeslutninger.
Kvalifikation: Beslutningstrin 1 afsluttes med et "ja", da MDSW er beregnet til at analysere medfødte data for at give information om disponering for en medicinsk tilstand eller sygdom og dermed opfylder kriterierne i IVDR, artikel 2, stk. 2, litra (b) og (c), der er anført i beslutningstrinnet. Da MDSW kun behandler data, der kommer fra medicinsk udstyr til in vitro-diagnostik, klassificeres softwaren som en IVD MDSW i henhold til trin 2.
Eksemplerne er kun vejledende for at illustrere, hvordan en bestemt regel kan være anvendt på et udstyr. Den angivne klassificering i eksemplet er ikke en bekræftelse på endelige klassificering af udstyret, da andre regler også kan overvejes. Samtidig skal det nævnes, at klassificering af udstyr i klasse IIa eller derover bør ske i dialog mellem fabrikanten og det bemyndigede organ.
Desuden afspejler den foreslåede klassifikation det specifikke erklærede formål eller sundhedssammenhængen eller situationen, hvor udstyret anvendes, som beskrevet i selve eksemplet. Enhver ændring af det erklærede formål eller sundhedssammenhæng / situation, hvor det samme udstyr bruges, kan resultere i en anden risikoklasse.
• MDSW beregnet til at udføre diagnose ved hjælp af billedanalyse til behandlingsbeslutninger hos patienter med akut slagtilfælde bør klassificeres som klasse III i henhold til regel 11 (a).
• Kognitiv terapi MDSW, der inkluderer en diagnostisk funktion, der er beregnet til at give feedback til softwaren for at bestemme opfølgningsterapi, fx software tilpasser behandling af depression baseret på diagnostisk feedback, bør være i klasse III i henhold til regel 22. Når en specialist bestemmer den nødvendige kognitive terapi baseret på resultatet leveret af MDSW, skal MDSW klassificeres som klasse IIa i henhold til regel 11 (a).
• Medicinsk udstyr herunder MDSW beregnet til kontinuerlig overvågning af vitale fysiologiske processer i anæstesi, intensiv pleje eller akut pleje bør klassificeres som klasse IIb jf. (regel 11 (b)).
• Medicinsk udstyr, herunder MDSW beregnet til at monitorere fysiologiske processer, der ikke anses for at være vitale, og udstyr beregnet til at blive brugt til at opnå aflæsninger af vitale fysiologiske signaler i rutinekontrol, herunder monitorering derhjemme, bør klassificeres som klasse IIa (regel 11 (b)).
• En mobilapp beregnet til at analysere en brugers hjerterytme, opdage anormaliteter og informere en læge bør klassificeres som klasse IIb i henhold til regel 11 (a), hvis oplysningerne leveret af softwaren er beregnet til at guide lægen i diagnosen.
• Diagnostisk MDSW beregnet til at score depression baseret på indtastede data om en patients symptomer (fx humør, angst) bør klassificeres somklasse IIb i henhold til regel 11 (a),
• Ambulante respiratoriske ventilationssystemer MDSW beregnet til langvarig brug (fx derhjemme), der advarer brugeren/operatøren om frakobling eller afvigelse fra det programmerede åndedræts volumen bør klassificeres som klasse IIb jf. regel 9.
• Aktivt udstyr, såsom elektroniske termometre og stetoskoper, som inkluderer MDSW beregnet til direkte diagnose, kan klassificeres som klasse IIa i henhold til regel 10 , tredje led siden kropstemperatur og puls betragtes som afgørende oplysninger til diagnose (implementeringsregel 3.7), hvor arten af variationerne i disse parametre ikke ville medføre øjeblikkelig fare for patienten.
• MDSW der har til formål at rangere terapeutiske forslag til sundhedspersonale baseret på patienthistorie, billedbehandlingstestresultater og patientkarakteristikker, for eksempel MDSW, der viser og rangerer alle tilgængelige kemoterapimuligheder for BRCA-positive individer, bør være klassificeret som klasse IIa i henhold til regel 11 (a)
• MDSW-app beregnet til at understøtte undfangelse ved at beregne brugerens fertilitetsstatus baseret på en valideret statistisk algoritme. Brugeren indtaster sundhedsdata inklusive basal kropstemperatur (BBT) og menstruationsdage for at spore og forudsige ægløsning. Den nuværende fertilitetsstatus for dagen reflekteres af en af tre indikatorlamper: rød (frugtbar), grøn (infertil) eller gul (indlæringsfase / cyklusudsving). Denne MDSW-app bør klassificeres som klasse I jf. Regel 11c.
• En softwareapp, der kan fjernjustere en hospitalsseng via Bluetooth, således at sengen indstilles efter sundhedspersonalets vurdering, som en del af patientens behandling, vil være kategoriseret som tilbehør til hospitalssengen, og derfor være klassificeret efter samme regel som hospitalssengen.
Klassificeringseksemplerne foroven er vejledende og har til formål at illustrere, hvordan en bestemt regel kan anvendes på et udstyr. Den angivne klassificering i eksemplerne foroven er ikke en bekræftelse af den endelige klassificering af udstyret, da andre regler også skal overvejes, og det i sidste ende vil være en konkret vurdering foretaget af fabrikanten og eventuelt i konsultation med et bemyndiget organ.
De væsentlige krav til medicinsk udstyr anses for opfyldt for udstyr, når de er i overensstemmelse med relevante eller dele af relevante harmoniserede standarder, hvis referencer er offentliggjort i Den Europæiske Unions Tidende. Referencer for nationale standarder offentliggøres for Danmarks vedkommende i publikationen Dansk Standard.
De harmoniserede standarder, der gælder for medicinsk udstyr, er et værktøj til at opfylde lovgivningens krav. Det er frivilligt at følge standarder, men de kan være en god hjælp i udviklingen af software og apps for at sikre kvalitet, funktionalitet og sikkerhed. Ud over de harmoniserede standarder for medicinsk udstyr kan der hentes værktøjer fra andre standarder, der vedrører software.
Liste over udvalgte standarder for medicinsk udstyr:
• EN ISO 15223-1:2016 Medical devices – Symbols to be used with medical device labels, labelling and information to be supplied – Part 1: General requirements
• EN ISO 13485:2016 Medical devices – Quality management systems – Requirements for regulatory purposes
• EN ISO 13485:2016/AC:2018 Medical devices – Quality management systems – Requirements for regulatory purposes
• EN ISO 14155:2020 Clinical investigation of medical devices for human subjects
• EN ISO 14971:2012 Medical Devices – Application of Risk Management to Medical Devices
• EN 60601-1:2006 Medical electrical equipment – Part 1: General requirements for basic safety and essential performance
• EN 60601-1-6:2010 Medical electrical equipment - Part 1-6: General requirements for basic safety and essential performance – Collateral Standard: Usability
• EN 62304:2015 Medical device software – Software life cycle processes
• EN 62366:2015 Medical devices – Application of usability engineering to medical devices
Andre relevante standarder for apps og software:
• CEN/TS 15260:2006 CEN Health informatics – Classification of safety risks from health informatics products
• ISO/IEC 20000-serien Information technology – Service management
• ISO/TS 25238:2007 ISO Health informatics – Classification of safety risks from health software
• ISO/TR 27809:2007 ISO Health informatics – Measures for ensuring patient safety of health software
• IEC/TR 80002:2009 Medical device software – Guidance on the application of ISO 14971 to medical device software
• IEC 82304:2016 Health software – Part 1: Requirements for product safety
• ISO/IEC 90003:2015 Software engineering – Guidelines for the application of ISO 9001:2000 to computer software
• Europa-Parlamentets og Rådets forordning (EU) 2017/745 af 5. april 2017 om medicinsk udstyr, om ændring af direktiv 2001/83/EF, forordning (EF) nr. 178/2002 og forordning (EF) nr. 1223/2009 og om ophævelse af Rådets direktiv 90/385/EØF og 93/42/EØF.
• Europa-Parlamentets og Rådets forordning (EU) 2017/746 af 5. april 2017 om medicinsk udstyr til in vitro-diagnostik og om ophævelse af direktiv 98/79/EF og Kommissions afgørelse 2010/227/EU.
• Lov om medicinsk udstyr, jf. lovbekendtgørelse nr. 139 af 15. februar 2016, som ændret ved § 3 i lov nr. 1687 af 26. december 2017, § 4 i lov nr. 1554 af 18. december 2018 og § 20 i lov nr. 1853 af 9. december 2020.
• Bekendtgørelse nr. 957 af 29. april 2021 om medicinsk udstyr og produkter uden medicinsk formål.
• MDCG 2019-11 Guidance on Qualification and Classification of Software - Regulation (EU) 2017/745 and Regulation (EU) 2017/746
• MDCG 2020-1 Guidance on clinical evaluation (MDR) / Performance evaluation (IVDR) of medical device software
• MDCG 2019.16 rev. 1 Guidance on cybersecurity for medical devices
• MDCG 2020-16 Guidance on Classification Rules for in vitro Diagnostic Medical Devices under Regulation (EU) 2017/746
Vejledninger fra EU kommissionens findes på denne hjemmeside: https://ec.europa.eu/health/md\_sector/new\_regulations/guidance\_en
Officielle noter
1 Artikel 2(1) i Forordning (EU) 2017/745 – MDR
2 Artikel 2(2) i Forordning (EU) 2017/746 – IVDR
3 Artikel 2(2) i Forordning (EU) 2017/745 – MDR
4 Artikel 2(4) i Forordning (EU) 2017/746 – IVDR
5 Artikel 32.2(c), Bilag I, Kapitel III Sektion 23.4(f), Bilag II Sektion 1.1(h) i Forordning (EU) 2017/745 – MDR Artikel 29.2(c) og Bilag II Sektion 1.1(m) i Forordning (EU) 2017/746 - IVDR
6 Artikel 2(4) i Forordning (EU) 2017/745 – MDR
7 Artikel 2(12) i Forordning (EU) 2017/745 – MDR
8 Artikel 2(12) i Forordning (EU) 2017/746 – IVDR
9 Artikel 2(28) i Forordning (EU) 2017/745 – MDR
10 Artikel 2(21) i Forordning (EU) 2017/746 – IVDR
11 Artikel 2(29) i Forordning (EU) 2017/745 – MDRArtikel 2(29) i Forordning (EU) 2017/745 – MDR
12 Artikel 2(22) i Forordning (EU) 2017/746 – IVDR
13 Artikel 2(41) og (42) i Forordning (EU) 2017/745 – MDR og artikel 2(33) og (34) i Forordning (EU) 2017/746 - IVDR
14 Artikel 2(1) i Forordning (EU) 2017/745 – MDR
15 Artikel 2(2) i Forordning (EU) 2017/746 – IVDR
16 I de tilfælde hvor et MDSW er tilsigtet anvendt af en lægmand, skal fabrikanten anvende sikkerheds- og ydeevnekrav som skitseret i MDR Bilag I. 22 og 23.4(w). eller IVDR Bilag I 9.4 og 20.4.2.
17 MDSW der kan betragtes som et IVD-udstyr til selvtest, skal betragtes som et udstyr som er tilsigtet anvendt af en lægmand.
18 Ifølge Artikel 2(2) Forordningen om medicinsk udstyr eller Forordningen om in vitro-diagnostisk medicinsk udstyr.
19 Kommunikation: Information der løber fra et punkt, kendt som kilden, til et andet punkt, modtageren. Kilde: IEEE 610.10-1994.
20 En kvalitativ tilgang har til formål at styre vægtningen af data. Vægtningen har til formål at vurdere bidraget fra hvert data op mod at opnå resultatet.
21 Se artikel 2 (53) i MDR for definition af kliniske fordele.
| 4.2 |
| Klassificeringsregler |
| 5. | Markedsføring og overensstemmelsesvurdering af MDSW |
| 5.1 | Mulighed 1: software, der er et medicinsk udstyr i sig selv |
| 5.2 | Mulighed 2: En integreret komponent / en del af et medicinsk udstyr/medicinsk udstyr til in vitro-diagnostik |
| 6. | Generelle principper for MDSW klinisk evaluering (MDR) / ydeevne evaluering (IVDR) processen |
| 6.1 | Introduktion |
| 6.2 | Beslutning af den valide kliniske sammenhæng / videnskabelige validitet |
| 6.3 | Teknisk ydeevne / Analytisk ydeevne |
| 6.4 | Klinisk ydeevne |
| 6.7 | Endelig analyse og konklusion den kliniske evaluering (MDR) / ydeevne evaluering (IVDR) |
| 6.8 | Løbende opdatering af den kliniske evaluering (MDR) / ydeevne evaluering (IVDR) |
| 7. | Moduler |
| 8. | Overvejelse af ændringer til et MDSW |
| 9. | Illustrerende eksempler på kvalifikation af software, der bruges i sundhedsvæsnet |
| 10. | Kvalifikationseksempler på medicinsk udstyrssoftware (MDSW) i henhold til Figur 1 og 2 |
| 11. | Klassificerings eksempler |
| 12. | Standarder |
| 13. | Liste over udvalgte standarder for medicinsk udstyr |
| 14. | Andre relevante standarder for apps og software |
| 15. | Lovgivning |
| 16. | Relaterede harmoniserede europæiske vejledninger |
